
Neuroblastoma (NB) is the most common extracranial solid cancer in childhood and the most common cancer in infancy, with an incidence of about six hundred and fifty cases per year in the U.S., and a hundred cases per year in the UK. Nearly half of neuroblastoma cases occur in children younger than two years. It is a neuroendocrine tumor, arising from any neural crest element of the sympathetic nervous system (SNS). It most frequently originates in one of the adrenal glands, but can also develop in nerve tissues in the neck, chest, abdomen, or pelvis.
Neuroblastoma is one of the few human malignancies known to demonstrate spontaneous regression from an undifferentiated state to a completely benign cellular appearance. It is a disease exhibiting extreme heterogeneity, and is stratified into three risk categories: low, intermediate, and high risk. Low-risk disease is most common in infants and good outcomes are common with observation only or surgery, whereas high-risk disease is difficult to treat successfully even with the most intensive multi-modal therapies available.
Esthesioneuroblastoma, also known as olfactory neuroblastoma, is believed to arise from the olfactory epithelium and its classification remains controversial. However, since it is not a sympathetic nervous system malignancy, esthesioneuroblastoma is a distinct clinical entity and is not to be confused with neuroblastoma.
Signs and symptoms

The first symptoms of neuroblastoma are often vague making diagnosis difficult. Fatigue, loss of appetite, fever, and joint pain are common. Symptoms depend on primary tumor locations and metastases if present:
- In the abdomen, a tumor may cause a swollen belly and constipation.
- A tumor in the chest may cause breathing problems.
- A tumor pressing on the spinal cord may cause weakness and thus an inability to stand, crawl, or walk.
- Bone lesions in the legs and hips may cause pain and limping.
- A tumor in the bones around the eyes or orbits may cause distinct bruising and swelling.
- Infiltration of the bone marrow may cause pallor from anemia.
Neuroblastoma often spreads to other parts of the body before any symptoms are apparent and 50 to 60% of all neuroblastoma cases present with metastases.
The most common location for neuroblastoma to originate (i.e., the primary tumor) is on the adrenal glands. This occurs in 40% of localized tumors and in 60% of cases of widespread disease. Neuroblastoma can also develop anywhere along the sympathetic nervous system chain from the neck to the pelvis. Frequencies in different locations include: neck (1%), chest (19%), abdomen (30% non-adrenal), or pelvis (1%). In rare cases, no primary tumor can be discerned.
Rare but characteristic presentations include transverse myelopathy (tumor spinal cord compression, 5% of cases), treatment-resistant diarrhea (tumor vasoactive intestinal peptide secretion, 4% of cases), Horner's syndrome (cervical tumor, 2.4% of cases), opsoclonus myoclonus syndrome and ataxia (suspected paraneoplastic cause, 1.3% of cases), and hypertension (catecholamine secretion or renal artery compression, 1.3% of cases).
Cause

The etiology of neuroblastoma is not well understood. The great majority of cases are sporadic and non-familial. About 1-2% of cases run in families and have been linked to specific gene mutations. Familial neuroblastoma in some cases is caused by rare germline mutations in the anaplastic lymphoma kinase (ALK) gene. Germline mutations in the PHOX2A or KIF1B gene have been implicated in familial neuroblastoma as well. Neuroblastoma is also a feature of neurofibromatosis type 1 and the Beckwith-Wiedemann syndrome.
MYCN oncogene amplification within the tumor is a common finding in neuroblastoma. The degree of amplification shows a bimodal distribution: either 3- to 10-fold, or 100- to 300-fold. The presence of this mutation is highly correlated to advanced stages of disease.
Duplicated segments of the LMO1 gene within neuroblastoma tumor cells have been shown to increase the risk of developing an aggressive form of the cancer.
Neuroblastoma has been linked to copy-number variation within the NBPF10 gene, which results in the 1q21.1 deletion syndrome or 1q21.1 duplication syndrome.
Several risk factors have been proposed and are the subject of ongoing research. Due to characteristic early onset many studies have focused on parental factors around conception and during gestation. Factors investigated have included occupation (i.e. exposure to chemicals in specific industries), smoking, alcohol consumption, use of medicinal drugs during pregnancy and birth factors; however, results have been inconclusive.
Other studies have examined possible links with atopy and exposure to infection early in life, use of hormones and fertility drugs, and maternal use of hair dye.
Diagnosis
The diagnosis is usually confirmed by a surgical pathologist, taking into account the clinical presentation, microscopic findings, and other laboratory tests.
Biochemistry
In about 90% of cases of neuroblastoma, elevated levels of catecholamines or their metabolites are found in the urine or blood. Catecholamines and their metabolites include dopamine, homovanillic acid (HVA), and/or vanillylmandelic acid (VMA).
Imaging
Another way to detect neuroblastoma is the mIBG scan (meta-iodobenzylguanidine), which is taken up by 90 to 95% of all neuroblastomas, often termed "mIBG-avid." The mechanism is that mIBG is taken up by sympathetic neurons, and is a functioning analog of the neurotransmitter norepinephrine. When it is radio-ionated with I-131 or I-123 (radioactive iodine isotopes), it is a very good radiopharmaceutical for diagnosis and monitoring of response to treatment for this disease. With a half-life of 13 hours, I-123 is the preferred isotope for imaging sensitivity and quality. I-131 has a half-life of 8 days and at higher doses is an effective therapy as targeted radiation against relapsed and refractory neuroblastoma.
Histology
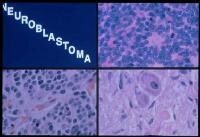
On microscopy, the tumor cells are typically described as small, round and blue, and rosette patterns (Homer-Wright rosettes) may be seen. Homer-Wright rosettes are tumor cells around neuropil, not to be confused with pseudorosettes which are tumor cells around a blood vessel. They are also distinct from the pseudorosettes of an ependymoma which consist of tumor cells with glial fibrillary acidic protein (GFAP)â€"positive processes tapering off toward a blood vessel (thus a combination of the two). A variety of immunohistochemical stains are used by pathologists to distinguish neuroblastomas from histological mimics, such as rhabdomyosarcoma, Ewing's sarcoma, lymphoma and Wilms' tumor.
Neuroblastoma is one of the peripheral neuroblastic tumors (pNTs) that have similar origins and show a wide pattern of differentiation ranging from benign ganglioneuroma to stroma-rich ganglioneuroblastoma with neuroblastic cells intermixed or in nodules, to highly malignant neuroblastoma. This distinction in the pre-treatment tumor pathology is an important prognostic factor, along with age and mitosis-karyorrhexis index (MKI). This pathology classification system describes "favorable" and "unfavorable" tumors by the International Neuroblastoma Pathology Committee (INPC, also called Shimada system) which was established in 1999 and revised in 2003.
Staging
The "International Neuroblastoma Staging System" (INSS) established in 1986 and revised in 1988 stratifies neuroblastoma according to its anatomical presence at diagnosis:
- Stage 1: Localized tumor confined to the area of origin.
- Stage 2A: Unilateral tumor with incomplete gross resection; identifiable ipsilateral and contralateral lymph node negative for tumor.
- Stage 2B: Unilateral tumor with complete or incomplete gross resection; with ipsilateral lymph node positive for tumor; identifiable contralateral lymph node negative for tumor.
- Stage 3: Tumor infiltrating across midline with or without regional lymph node involvement; or unilateral tumor with contralateral lymph node involvement; or midline tumor with bilateral lymph node involvement.
- Stage 4: Dissemination of tumor to distant lymph nodes, bone marrow, bone, liver, or other organs except as defined by Stage 4S.
- Stage 4S: Age <1 year old with localized primary tumor as defined in Stage 1 or 2, with dissemination limited to liver, skin, or bone marrow (less than 10 percent of nucleated bone marrow cells are tumors).
Although international agreement on staging (INSS) has been used, the need for an international consensus on risk assignment has also been recognized in order to compare similar cohorts in results of studies. Beginning in 2005, representatives of the major pediatric oncology cooperative groups have met to review data for 8,800 neuroblastoma patients treated in Europe, Japan, USA, Canada, and Australia between 1990 and 2002. This task force has proposed the International Neuroblastoma Risk Group (INRG) classification system. Retrospective studies revealed the high survival rate of 12-18 month old age group, previously categorized as high-risk, and prompted the decision to reclassify 12-18 month old children without N-myc (also commonly referred to as MYCN) amplification to intermediate risk category.
The new INRG risk assignment will classify neuroblastoma at diagnosis based on a new International Neuroblastoma Risk Group Staging System (INRGSS):
- Stage L1: Localized disease without image-defined risk factors.
- Stage L2: Localized disease with image-defined risk factors.
- Stage M: Metastatic disease.
- Stage MS: Metastatic disease "special" where MS is equivalent to stage 4S.
The new risk stratification will be based on the new INRGSS staging system, age (dichotomized at 18 months), tumor grade, N-myc amplification, unbalanced 11q aberration, and ploidy into four pre-treatment risk groups: very low, low, intermediate, and high risk.
Screening
Urine catecholamine level can be elevated in pre-clinical neuroblastoma. Screening asymptomatic infants at three weeks, six months, and one year has been performed in Japan, Canada, Austria and Germany since the 1980s. Japan began screening six-month olds for neuroblastoma via analysis of the levels of homovanillic acid and vanilmandelic acid in 1984. Screening was halted in 2004 after studies in Canada and Germany showed no reduction in deaths due to neuroblastoma, but rather caused an increase in diagnoses that would have disappeared without treatment, subjecting those infants to unnecessary surgery and chemotherapy.
Treatment

When the lesion is localized, it is generally curable. However, long-term survival for children with advanced disease older than 18 months of age is poor despite aggressive multimodal therapy (intensive chemotherapy, surgery, radiation therapy, stem cell transplant, differentiation agent isotretinoin also called 13-cis-retinoic acid, and frequently immunotherapy with anti-GD2 monoclonal antibody therapy).
Biologic and genetic characteristics have been identified, which, when added to classic clinical staging, has allowed patient assignment to risk groups for planning treatment intensity. These criteria include the age of the patient, extent of disease spread, microscopic appearance, and genetic features including DNA ploidy and N-myc oncogene amplification (N-myc regulates microRNAs), into low, intermediate, and high risk disease. A recent biology study (COG ANBL00B1) analyzed 2687 neuroblastoma patients and the spectrum of risk assignment was determined: 37% of neuroblastoma cases are low risk, 18% are intermediate risk, and 45% are high risk. (There is some evidence that the high- and low-risk types are caused by different mechanisms, and are not merely two different degrees of expression of the same mechanism.)
The therapies for these different risk categories are very different.
- Low-risk disease can frequently be observed without any treatment at all or cured with surgery alone.
- Intermediate-risk disease is treated with surgery and chemotherapy.
- High-risk neuroblastoma is treated with intensive chemotherapy, surgery, radiation therapy, bone marrow / hematopoietic stem cell transplantation, biological-based therapy with 13-cis-retinoic acid (isotretinoin or Accutane) and antibody therapy usually administered with the cytokines GM-CSF and IL-2.
With current treatments, patients with low and intermediate risk disease have an excellent prognosis with cure rates above 90% for low risk and 70%-90% for intermediate risk. In contrast, therapy for high-risk neuroblastoma the past two decades resulted in cures only about 30% of the time. The addition of antibody therapy has raised survival rates for high-risk disease significantly. In March 2009 an early analysis of a Children's Oncology Group (COG) study with 226 high-risk patients showed that two years after stem cell transplant 66% of the group randomized to received ch14.18 antibody with GM-CSF and IL-2 were alive and disease-free compared to only 46% in the group that did not receive the antibody. The randomization was stopped so all patients enrolling on the trial will receive the antibody therapy.
Chemotherapy agents used in combination have been found to be effective against neuroblastoma. Agents commonly used in induction and for stem cell transplant conditioning are platinum compounds (cisplatin, carboplatin), alkylating agents (cyclophosphamide, ifosfamide, melphalan), topoisomerase II inhibitor (etoposide), anthracycline antibiotics (doxorubicin) and vinca alkaloids (vincristine). Some newer regimens include topoisomerase I inhibitors (topotecan and irinotecan) in induction which have been found to be effective against recurrent disease.
Prognosis

Between 20% and 50% of high-risk cases do not respond adequately to induction high-dose chemotherapy and are progressive or refractory. Relapse after completion of frontline therapy is also common. Further treatment is available in phase I and phase II clinical trials that test new agents and combinations of agents against neuroblastoma, but the outcome remains very poor for relapsed high-risk disease.
Most long-term survivors alive today had low or intermediate risk disease and milder courses of treatment compared to high-risk disease. The majority of survivors have long-term effects from the treatment. Survivors of intermediate and high-risk treatment often experience hearing loss. Growth reduction, thyroid function disorders, learning difficulties, and greater risk of secondary cancers affect survivors of high-risk disease. An estimated two of three survivors of childhood cancer will ultimately develop at least one chronic and sometimes life-threatening health problem within 20 to 30 years after the cancer diagnosis.
Cytogenetic profiles
Based on a series of 493 neuroblastoma samples, it has been reported that overall genomic pattern, as tested by array-based karyotyping, is a predictor of outcome in neuroblastoma:
- Tumors presenting exclusively with whole chromosome copy number changes were associated with excellent survival.
- Tumors presenting with any kind of segmental chromosome copy number changes were associated with a high risk of relapse.
- Within tumors showing segmental alterations, additional independent predictors of decreased overall survival were N-myc amplification, 1p and 11q deletions, and 1q gain.
Earlier publications categorized neuroblastomas into three major subtypes based on cytogenetic profiles:
- Subtype 1: favorable neuroblastoma with near triploidy and a predominance of numerical gains and losses, mostly representing non-metastatic NB stages 1, 2 and 4S.
- Subtypes 2A and 2B: found in unfavorable widespread neuroblastoma, stages 3 and 4, with 11q loss and 17q gain without N-myc amplification (subtype 2A) or with N-myc amplification often together with 1p deletions and 17q gain (subtype 2B).
Virtual karyotyping can be performed on fresh or paraffin-embedded tumors to assess copy number at these loci. SNP array virtual karyotyping is preferred for tumor samples, including neuroblastomas, because they can detect copy neutral loss of heterozygosity (acquired uniparental disomy). Copy neutral LOH can be biologically equivalent to a deletion and has been detected at key loci in neuroblastoma. ArrayCGH, FISH, or conventional cytogenetics cannot detect copy neutral LOH.
Epidemiology

Neuroblastoma comprises 6-10% of all childhood cancers, and 15% of cancer deaths in children. The annual mortality rate is 10 per million children in the 0- to 4-year-old age group, and 4 per million in the 4- to 9-year old age group.
The highest incidence is in the first year of life, and some cases are congenital. The age range is broad, including older children and adults, but only 10% of cases occur in people older than 5 years of age. A large European study reported less than 2% of over 4000 neuroblastoma cases were over 18 years old.
History

In 1864 German physician Rudolf Virchow was the first to describe an abdominal tumor in a child as a "glioma". The characteristics of tumors from the sympathetic nervous system and the adrenal medulla were then noted in 1891 by German pathologist Felix Marchand. In 1901 the distinctive presentation of stage 4S in infants (liver but no bone metastases) was described by William Pepper. In 1910 James Homer Wright understood the tumor to originate from primitive neural cells, and named it neuroblastoma. He also noted the circular clumps of cells in bone marrow samples which are now termed "Homer-Wright pseudorosettes".
Research
Treatments
Recent focus has been to reduce therapy for low and intermediate risk neuroblastoma while maintaining survival rates at 90%. A study of 467 intermediate risk patients enrolled in A3961 from 1997 to 2005 confirmed the hypothesis that therapy could be successfully reduced for this risk group. Those with favorable characteristics (tumor grade and response) received four cycles of chemotherapy, and those with unfavorable characteristics received eight cycles, with three-year event free survival and overall survival stable at 90% for the entire cohort. Future plans are to intensify treatment for those patients with aberration of 1p36 or 11q23 chromosomes as well as for those who lack early response to treatment.
By contrast, focus the past 20 years or more has been to intensify treatment for high-risk neuroblastoma. Chemotherapy induction variations, timing of surgery, stem cell transplant regimens, various delivery schemes for radiation, and use of monoclonal antibodies and retinoids to treat minimal residual disease continue to be examined. Recent phase III clinical trials with randomization have been carried out to answer these questions to improve survival of high-risk disease:
- 1982-1985: European Neuroblastoma Study Group (ENSG1) enrolled 167 children and randomized to melphalan autologous bone marrow transplant or no further therapy (no radiation therapy given to any). Transplant and no-transplant arms each had 65 patients, and recent long-term follow-up report revealed significantly better 5 year event-free survival for stage 4 over 1 year old in melphalan-transplant group versus no further treatment: 33% versus 17% respectively.
- 1990-1999: European study (EU-20592 or CCLGNB-1990-11) randomized 262 high-risk children over 1 year old and revealed higher survival rate for rapid sequence induction (10-day cycle) versus standard induction (21-day cycle) with same total dose. Ten-year event free survival was 27% and 18% respectively with non-aggressive surgical approach, no radiotherapy, and melphalan-only autologous bone marrow or stem cell transplant for both groups.
- 1991-1996: Phase III trial with two sequential randomizations for 379 high-risk NB patients was carried out by the Children's Cancer Group (CCG-3891) which demonstrated improved survival with myeloablative therapy (with total body irradiation) and 13-cis-retinoic acid (Accutane) with 50 patients in each of the four arms of the study.,
- 1996-2003: The German (GPOH) study NB97 compared outcomes of 295 high-risk NB patients randomized for stem cell transplant or consolidation chemotherapy. Results showed increased survival with transplant.
- 2000-2006: The recent study (COG-A3973) questioned the need for purged stem cells for CEM-LI (carboplatin, etoposide, melphalan, with local irradiation) transplant, and accrued 486 patients. Purging stem cells was not found to improve survival
- 2000-2012: A concurrent study (COG-ANBL0032) determined in early review that the antibody ch14.18 with interleukin 2 and GMCSF (studied retrospectively in German GPOH NB90 and NB 97 at a lower dose and without cytokines) improved survival, and will accrue a total of 423 patients. A follow on Phase III study COG-ANBL0931 opened Jan 2010 to accrue 105 patients to gather further safety and efficacy data for FDA approval.
- 2002-2008: SIOP (International Society of Paediatric Oncology) formed the European SIOP Neuroblastoma Group (SIOPEN) in 1994 and activated a phase III high-risk NB protocol in 2002 (SIOP-EUROPE-HR-NBL-1) using "rapid" COJEC (8 cycles of chemotherapy given at 10-day intervals) followed by transplant randomization to CEM (carboplatin, etoposide, melphalan) or BuMel (busulfan, melphalan) and the study has been recently amended to randomize children to ch14.18 antibody treatment with or without subcutaneous IL2 (without GM-CSF as given in the COG). This study recently reported the benefit of growth factors (GCSF), and all patients receive retinoic acid. This trial will accrue 1000 patients (175 per year).
- 2005-2010: The current German NB2004 randomization will include MIBG therapy and randomize topotecan use in up-front therapy and will accrue a total of 642 for all risk groups (roughly half will be high-risk). After transplant, the high-risk protocol includes six months of cis-retinoic acid, a three-month break, and another three months of retinoic acid.
- 2007: The COG phase III ANBL0532 trial opened December 2007 for accrual of 495 and will compare single versus tandem transplants, and induction begins with two cycles of topotecan.
In addition to these phase III studies, some research institutions offer pilot treatment protocols. For example, St Jude's recently finished (2007) testing a new up-front chemotherapy regimen in 23 children which included irinotecan and gefitinib with 16 months of maintenance chemotherapy after stem cell transplant with alternating oral 13-cis-retinoic acid and topotecan. Memorial Sloan-Kettering Cancer Center in New York offers treatment that includes a mouse-derived monoclonal antibody, 3F8, used in protocols since the mid-1980s. This antibody is used for treating minimal residual disease or consolidation instead of stem cell transplant. A new pilot protocol COG-ANBL09P1 available for newly diagnosed (high-risk) children at several Children's Oncology Group (COG) centers will offer MIBG radiotherapy and chemotherapy for the transplant regimen.
Refractory and relapsed neuroblastoma
Some children (particularly in high-risk cases) do not respond completely to frontline treatment (with a complete response or very good partial response) and are labeled refractory. These children are removed from the frontline therapy (clinical trial) and are eligible for clinical trials using new therapies. Many high-risk children have a good response to frontline therapy and achieve a remission, but later the disease recurs (relapse). These children are also eligible for new therapies being tested in clinical trials.
Chemotherapy with topotecan and cyclophosphamide is frequently used in refractory setting and after relapse. A randomized study (2004) with 119 patients (comparing topotecan alone to topotecan and cyclophosphamide) revealed a 31% complete or partial response rate with two-year progression-free survival at 36% in the topotecan and cyclophosphamide group. Irinotecan (intravenous or oral) and oral temozolomide are also used in refractory and recurrent neuroblastoma.
Many phase I and phase II trials are currently testing new agents against neuroblastoma in children who have relapsed or are resistant to initial therapy. Investigators are currently studying new agents, alone and in new combinations, using small molecule targeted therapy, 131-I MIBG radiation therapy, angiogenesis agents, new monoclonal antibodies, vaccines, oncolytic viruses, as well as new myeloablative regimens.
A group of 16 children's hospitals in the United States known as the New Advances in Neuroblastoma Therapy (NANT) consortium coordinates the I-131 MIBG radiation therapy trials. The NANT consortium also offers trials using an oral powder formulation of fenretinide, intravenous fenretinide, bisphosphonate (Zometa) with other agents, and combining I-131 MIBG with the inhibitor vorinostat.
Other research study groups such as The Neuroblastoma and Medulloblastoma Translational Research Consortium (NMTRC) also conduct clinical trials to treat relapse neuroblastoma. Institutions in Europe are studying novel therapies to treat relapse, including haploidentical stem cell transplant. Many hospitals conduct their own institutional studies as well.
The protein p53 is believed to play a role in the development of resistance to chemotherapy. A November 2009 study in mice shows that activating the tumor suppressor p53 with a new drug, nutlin-3, may slow tumor growth. In this study, physician Tom Van Maerken of Ghent University Hospital in Belgium and his colleagues used nutlin-3 to neutralize MDM2, a protein that binds to the p53 protein and obstructs p53's ability to trigger programmed cell death. Earlier studies have shown that nutlin-3 can specifically prevent MDM2 from disabling p53.
Legislative efforts
U.S. Representative Chet Edwards of Waco, Texas, successfully introduced legislation to earmark $150 million toward a cure for neuroblastoma and other cancers. The measure was signed into law in July 2008 by U.S. President George W. Bush. Edwards was inspired in the endeavor by the illness and subsequent death of Erin Channing Buenger (1997â€"2009) of Bryan, daughter of one of his constituents, Walter L. Buenger, head of the history department at Texas A&M University.
Fundraising
Several organizations fundraise for research into neuroblastoma. Tom Hanks is the Honorary Patron of the James Fund, a leading fundraising organization in support of clinical research. On December 31, 2014, SickKids Foundation takes over stewardship of The James Funds. The Cincinnati Bengals announced in 2014 that they would donate proceeds from the jersey sales of defensive lineman Devon Still's jersey to the Cincinnati Children's Hospital, and will present a check from more than $1.3 million to Still's daughter Leah during Thursday night's game on November 6, 2014. Cincinnati Bengals defensive lineman Devon Still's jersey is the 11th-most popular in the NFL
References
External links
- VIDEO - Neuroblastoma: New Advances in the Treatment of a Deadly Disease Dr. Kenneth DeSantes speaks at the University of Wisconsin-Madison School of Medicine and Public Health. 2011.
- Neuroblastoma at DMOZ





0 comments:
Post a Comment