
Adrenoleukodystrophy (also known as X-linked adrenoleukodystrophy, ALD, X-ALD, adrenomyeloneuropathy, AMN, Siemerlingâ€"Creutzfeldt disease or bronze Schilder disease) is a disorder of peroxisomal fatty acid beta oxidation which results in the accumulation of very-long chain fatty acids in tissues throughout the body. The most severely affected tissues are the myelin in the central nervous system, the adrenal cortex and the Leydig cells in the testes. Clinically, ALD is a heterogenous disorder, presenting with several distinct phenotypes, and no clear pattern of genotype-phenotype correlation. As an X-linked disorder, ALD presents most commonly in males, however approximately 50% of heterozygote females show some symptoms later in life. Approximately two-thirds of ALD patients will present with the childhood cerebral form of the disease, which is the most severe form. It is characterized by normal development in early childhood, followed by rapid degeneration to a vegetative state. The other forms of ALD vary in terms of onset and clinical severity, ranging from adrenal insufficiency to progressive paraparesis in early adulthood (this form of the disease is typically known as adrenomyeloneuropathy).
ALD is caused by mutations in ABCD1, a gene located on the X chromosome that codes for ALD, a peroxisomal membrane transporter protein. The exact mechanism of the pathogenesis of the various forms of ALD is not known. Biochemically, individuals with ALD show very high levels of unbranched, saturated, very long chain fatty acids, particularly cerotic acid (26:0). The level of cerotic acid in plasma does not correlate with clinical presentation. Treatment options for ALD are limited. Dietary treatment is with Lorenzo's oil. For the childhood cerebral form, stem cell transplant and gene therapy are options if the disease is detected early in the clinical course. Adrenal insufficiency in ALD patients can be successfully treated. ALD is the most common peroxisomal inborn error of metabolism, with an incidence estimated between 1:20,000 and 1:50,000. It does not have a significantly higher incidence in any specific ethnic groups.
Clinical presentation
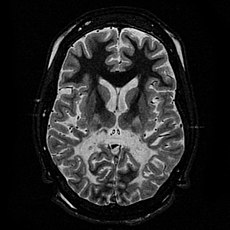
ALD is a clinically heterogeneous disease. The different clinical presentations are complicated by the pattern of X-linked recessive inheritance. There have been seven phenotypes described in male patients with ABCD1 mutations and five in females. Initial symptoms in boys affected with the childhood cerebral form of ALD include emotional instability, hyperactivity and disruptive behavior at school. Older patients affected with the cerebral form will present with similar symptoms. Untreated, cerebral ALD is characterized by progressive demyelination leading to a vegetative state and death. Adult males with an adrenomyeloneuropathy presentation typically present initially with muscle stiffness, paraparesis and sexual dysfunction. All patients with clinically recognized ALD phenotypes are at risk for adrenal insufficiency. There is no reliable way to predict which form of the disease an affected individual will develop, with multiple phenotypes being demonstrated within families. Onset of adrenal insufficiency is often the first symptom, appearing as early as two years of age.
Male adrenoleukodystrophy phenotypes
Female adrenoleukodystrophy phenotypes
Diagnosis
The clinical presentation of ALD can vary greatly, making diagnosis difficult. With the variety of phenotypes, clinical suspicion of ALD can result from a variety of different presentations. Symptoms vary based on the disease phenotype, and even within families or between twins. When ALD is suspected based on clinical symptoms, the initial testing usually includes plasma very long chain fatty acid (VLCFA) determination using gas chromatography-mass spectrometry. The concentration of unsaturated VLCFA, particularly 26 carbon chains are significantly elevated in males with ALD, even prior to the development of other symptoms. Confirmation of ALD after positive plasma VLCFA determination usually involves molecular genetic analysis of ABCD1. In females, where plasma VLCFA measurement is not always conclusive (some female carriers will have normal VLCFA in plasma), molecular analysis is preferred, particularly in cases where the mutation in the family is known. Although the clinical phenotype is highly variable among affected males, the elevations of VLCFA are present in all males with an ABCD1 mutation.
Because the characteristic elevations associated with ALD are present at birth, well before any symptoms are apparent, there have been methods developed in the interests of including it in newborn screening programs. One of the difficulties with ALD as a disease included in universal newborn screening is the difficulty in predicting the eventual phenotype that an individual will express. The accepted treatment for affected boys presenting with the cerebral childhood form of the disease is a bone marrow transplant, a procedure which carries significant risks. However, because most affected males will demonstrate adrenal insufficiency, early discovery and treatment of this symptom could potentially prevent complications and allow these patients to be monitored for other treatment in the future, depending on the progression of their disease.
The Loes score is a rating of the severity of abnormalities in the brain found on MRI. It ranges from 0 to 34, based on a point system derived from the location and extent of disease and the presence of atrophy in the brain, either localized to specific points or generally throughout the brain. A Loes score of 0.5 or less is classified as normal, while a Loes score of 14 or greater is considered severe. It was developed by neuroradiologist Daniel J. Loes MD and is an important tool in assessing disease progression and the effectiveness of therapy.
Genetics
ALD is caused by mutations in ABCD1, located at Xq28 and demonstrates X-linked recessive inheritance. The gene ABCD1 encodes a peroxisomal membrane transporter which is responsible for transporting very long chain fatty acid substrate into the peroxisomes for degradation. Mutations in this gene that interfere with this process cause this syndrome.
Males with an ABCD1 mutation are hemizygous, as they only have a single X chromosome. Female carriers will typically avoid the most severe manifestations of the disease, but often become symptomatic later in life. Although, the detection of an ABCD1 mutation identifies an individual who is affected with a form of ALD, however there is no genotype - phenotype correlation. Within a family, there will often be several different phenotypes, despite the presence of the same causative mutation. In one case, a family with six affected members displayed five different phenotypes. There are no common mutations that cause ALD, most are private or familial. Almost 600 different mutations have been identified, approximately half are missense mutations, one quarter are frameshifts, with in-frame deletions and splicing defects making up the remainder. The incidence of new mutations in ALD (those occurring spontaneously, rather than being inherited from a carrier parent) is estimated at approximately 4.1%, with the possibility that these are due to germline mosaicism.
Pathogenesis
The exact cause for the varied collection of symptoms found in the different ALD phenotypes is not clear. The white matter of the brain, the Leydig cells of the testes and the adrenal cortex are the most severely affected systems. The excess VLCFA can be detected in almost all tissues of the body, despite the localization of symptoms. Successful treatment of the demyelination process that affects the brain with either stem cell transplant or gene therapy does not immediately normalize the VLCFA levels in body tissues. The levels of VLCFA can be normalized by treatment with Lorenzo's oil, but this does not alter the progression of the disease. It is unclear whether the accumulation of VLCFA is associated with the pathogenesis of the disease in a specific way, or if it is a biochemical phenotype, useful for identification.
Treatment

Dietary therapy
Initial attempts at dietary therapy in ALD involved restricting the intake of very-long chain fatty acids (VLCFA). Dietary intake is not the only source for VLCFA in the body, as they are also synthesized endogenously. This dietary restriction did not impact the levels of VLCFA in plasma and other body tissues. After the realization that endogenous synthesis was an important contribution to VLCFA in the body, efforts at dietary therapy shifted to inhibiting these synthetic pathways in the body. The parents of Lorenzo Odone, a boy with ALD, spearheaded efforts to develop a dietary treatment to slow the progression of the disease. They developed a mixture of unsaturated fatty acids (glycerol trioleate and glyceryl trierucate in a 4:1 ratio), known as Lorenzo's oil that inhibits elongation of saturated fatty acids in the body. Supplementation with Lorenzo's oil has been found to normalize the VLCFA concentrations in the body, although its effectiveness at treating the cerebral manifestations of the disease are still controversial and unproven. Trials with Lorenzo's oil have shown that it does not stop the neurological degradation in symptomatic patients, nor does it improve adrenal function.
Transplant
While dietary therapy has been shown to be effective to normalize the very-long chain fatty acid concentrations in the plasma of individuals with ALD, allogeneic hematopoietic stem cell transplants are the only treatment that can stop the demyelination that is the hallmark of the cerebral forms of the disease. In order to be effective, the transplant must be done at an early stage of the disease; if the demyelination has progressed, transplant can worsen the outcome, and increase the rate of decline. While transplants have been shown to be effective at halting the demyelination process in those presenting with the childhood cerebral form of ALD, follow-up of these patients has shown that it does not improve adrenal function.
Gene therapy
For patients where an appropriate match for a transplant cannot be found, there have been investigations into the use of gene therapy. Appropriate vectors are selected and modified to express wild type ABCD1, which is then transplanted into the patients using a similar procedure as for a bone marrow or stem cell transplant. Gene therapy has only been tried on a small number of patients, mainly in France. These patients were only considered for gene therapy after there was no HLA match for a traditional transplant. In two reported cases, the gene therapy was successful, with a resolution of the demyelination process up to two years after the procedure. Although the gene therapy was successful in resolving the neurological symptoms, plasma VLCFA levels remained elevated.
Adrenal insufficiency
Treatment of the adrenal insufficiency that can accompany any of the common male phenotypes of ALD does not resolve any of the neurological symptoms. Hormone replacement is standard for ALD patients demonstrating adrenal insufficiency. Adrenal insufficiency does not resolve with successful transplant; most patients still require hormone replacement.
Incidence
ALD has not been shown to have an increased incidence in any specific country or ethnic group. In the United States, the incidence of affected males is estimated at 1:21,000. Overall incidence of hemizygous males and carrier females is estimated at 1:16,800. The reported incidence in France is estimated at 1:22,000.
References
External links

- ALD Life
- European Leukodystrophy Foundation
- Fight ALD
- March of Dimes Foundation
- United Leukodystrophy Foundation
- Adrenoleukodystrophy at DMOZ
- adrenoleukodystrophy at NINDS
- Images of ALD at USUHS
- Adrenoleukodystrophy at National Center for Biotechnology Information






I'm 61 years old, I contracted hpv in 2011' I has be taking lot treatment for it and embarrassedsome months ago the wart stated coming out seriously, I used lot recommendation because there was lot warts around my anus and was so . but today I'm totally happy I got the virus eliminated by using natural treatment from Dr Onokun herbal center after his treatment I got cured. all the warts went away' seriously believed Dr Onokun he have the cure for human papillomavirus because he has eliminated hpv been in my body since 2011, Dr Onokun make it possible for me. Here is Dr Onokun email to reach him: Dronokunherbalcure@gmail.com he is welled capable of curing terrible diseases.
ReplyDelete