
DNA methylation is a biochemical process where a methyl group is added to the cytosine or adenine DNA nucleotides. The rate of cytosine DNA methylation differs strongly between species, e.g. absolute quantification by mass spectrometry revealed 14% of cytosines methylated in Arabidopsis thaliana, 8% in Mus musculus, 2.3% in Escherichia coli, 0.03% in Drosophila, and virtually none (< 0.0002%) in yeast species. DNA methylation may stably alter the expression of genes in cells as cells divide and differentiate from embryonic stem cells into specific tissues. The resulting change is normally permanent and unidirectional, preventing a cell from reverting to a stem cell or converting into a different cell type. DNA methylation is typically removed during zygote formation and re-established through successive cell divisions during development. However, the latest research shows that hydroxylation of methyl groups occurs rather than complete removal of methyl groups in the zygote. Some methylation modifications that regulate gene expression are heritable and cause genomic imprinting.
DNA methylation suppresses the expression of endogenous retroviral genes and other harmful stretches of DNA that have been incorporated into the host genome over time. DNA methylation also forms the basis of chromatin structure, which enables a single cell to grow into multiple organs or perform multiple functions. DNA methylation also plays a crucial role in the development of nearly all types of cancer.
DNA methylation at the 5 position of cytosine has the specific effect of reducing gene expression and has been found in every vertebrate examined. In adult somatic cells (cells in the body, not used for reproduction), DNA methylation typically occurs in a CpG dinucleotide context; non-CpG methylation is prevalent in embryonic stem cells, and has also been indicated in neural development.
In mammals
DNA methylation is essential for normal development and is associated with a number of key processes including genomic imprinting, X-chromosome inactivation, suppression of repetitive elements, and carcinogenesis.
Between 60% and 90% of all CpGs are methylated in mammals. Methylated C residues spontaneously deaminate to form T residues over time; hence CpG dinucleotides steadily deaminate to TpG dinucleotides, which is evidenced by the under-representation of CpG dinucleotides in the human genome (they occur at only 21% of the expected frequency). (On the other hand, spontaneous deamination of unmethylated C residues gives rise to U residues, a change that is quickly recognized and repaired by the cell.)
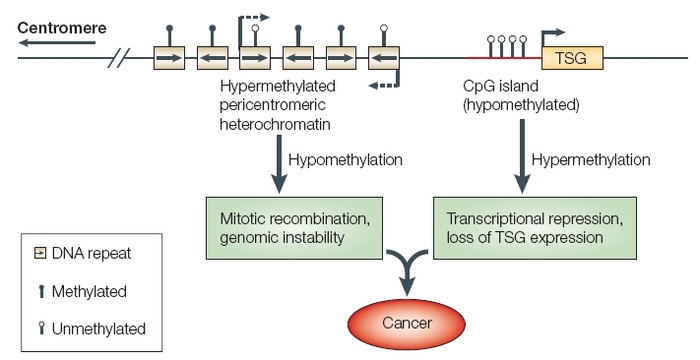
Unmethylated CpGs are often grouped in clusters called CpG islands, which are present in the 5' regulatory regions of many genes. In many disease processes, such as cancer, gene promoter CpG islands acquire abnormal hypermethylation, which results in transcriptional silencing that can be inherited by daughter cells following cell division. Alterations of DNA methylation have been recognized as an important component of cancer development. Hypomethylation, in general, arises earlier and is linked to chromosomal instability and loss of imprinting, whereas hypermethylation is associated with promoters and can arise secondary to gene (oncogene suppressor) silencing, but might be a target for epigenetic therapy.
DNA methylation may affect the transcription of genes in two ways. First, the methylation of DNA itself may physically impede the binding of transcriptional proteins to the gene, and second, and likely more important, methylated DNA may be bound by proteins known as methyl-CpG-binding domain proteins (MBDs). MBD proteins then recruit additional proteins to the locus, such as histone deacetylases and other chromatin remodeling proteins that can modify histones, thereby forming compact, inactive chromatin, termed heterochromatin. This link between DNA methylation and chromatin structure is very important. In particular, loss of methyl-CpG-binding protein 2 (MeCP2) has been implicated in Rett syndrome; and methyl-CpG-binding domain protein 2 (MBD2) mediates the transcriptional silencing of hypermethylated genes in cancer.
Research has suggested that long-term memory storage in humans may be regulated by DNA methylation.
DNA methylation levels can be used to estimate age, forming an accurate biological clock in humans and chimpanzees.
In cancer
DNA methylation is an important regulator of gene transcription and a large body of evidence has demonstrated that genes with high levels of 5-methylcytosine in their promoter region are transcriptionally silent, and that DNA methylation gradually accumulates upon long-term gene silencing. DNA methylation is essential during embryonic development, and in somatic cells, patterns of DNA methylation are generally transmitted to daughter cells with a high fidelity. Aberrant DNA methylation patterns â€" hypermethylation and hypomethylation compared to normal tissue â€" have been associated with a large number of human malignancies. Hypermethylation typically occurs at CpG islands in the promoter region and is associated with gene inactivation. A lower level of leukocyte DNA methylation is associated with many types of cancer. Global hypomethylation has also been implicated in the development and progression of cancer through different mechanisms. Typically, there is hypermethylation of tumor suppressor genes and hypomethylation of oncogenes.
In atherosclerosis
Epigenetic modifications such as DNA methylation have been implicated in cardiovascular disease, including atherosclerosis. In animal models of atherosclerosis, vascular tissue as well as blood cells such as mononuclear blood cells exhibit global hypomethylation with gene-specific areas of hypermethylation. DNA methylation polymorphisms may be used as an early biomarker of atherosclerosis since they are present before lesions are observed, which may provide an early tool for detection and risk prevention.
Two of the cell types targeted for DNA methylation polymorphisms are monocytes and lymphocytes, which experience an overall hypomethylation. One proposed mechanism behind this global hypomethylation is elevated homocysteine levels causing hyperhomocysteinemia, a known risk factor for cardiovascular disease. High plasma levels of homocysteine inhibit DNA methyltransferases, which causes hypomethylation. Hypomethylation of DNA affects gene that alter smooth muscle cell proliferation, cause endothelial cell dysfunction, and increase inflammatory mediators, all of which are critical in forming atherosclerotic lesions. High levels of homocysteine also result in hypermethylation of CpG islands in the promoter region of the estrogen receptor alpha (ERα) gene, causing its down regulation. ERα protects against atherosclerosis due to its action as a growth suppressor, causing the smooth muscle cells to remain in a quiescent state. Hypermethylation of the ERα promoter thus allows intimal smooth muscle cells to proliferate excessively and contribute to the development of the atherosclerotic lesion.
Another gene that experiences a change in methylation status in atherosclerosis is the monocarboxylate transporter (MCT3), which produces a protein responsible for the transport of lactate and other ketone bodies out of many cell types, including vascular smooth muscle cells. In atherosclerosis patients, there is an increase in methylation of the CpG islands in exon 2, which decreases MCT3 protein expression. The down regulation of MCT3 impairs lactate transport, and significantly increases smooth muscle cell proliferation, which further contributes to the atherosclerotic lesion. An ex vivo experiment using the demethylating agent Decitabine (5-aza-2 -deoxycytidine) was shown to induce MCT3 expression in a dose dependant manner, as all hypermethylated sites in the exon 2 CpG island became demethylated after treatment. This may serve as a novel therapeutic agent to treat atherosclerosis, although no human studies have been conducted thus far.
In aging
A longitudinal study of twin children, showed that between the ages of 5 and 10 there was divergence of methylation patterns due to environmental rather than genetic influences. There is a global loss of DNA methylation during aging. But some genes become hypermethylated with age, including genes for the estrogen receptor, p16, and insulin-like growth factor 2. Biological clocks such as an epigenetic clock, are promising biomarkers of aging.
In exercise
High intensity exercise has been shown to result in reduced DNA methylation in skeletal muscle. Promoter methylation of PGC-1α and PDK4 were immediately reduced after high intensity exercise, whereas PPAR-γ methylation was not reduced until three hours after exercise. By contrast, six months of exercise in previously sedentary middle-age men resulted in increased methylation in adipose tissue. One study showed a possible increase in global genomic DNA methylation of white blood cells with more physical activity in non-Hispanics.
DNA methyltransferases
In mammalian cells, DNA methylation occurs mainly at the C5 position of CpG dinucleotides and is carried out by two general classes of enzymatic activities â€" maintenance methylation and de novo methylation.
Maintenance methylation activity is necessary to preserve DNA methylation after every cellular DNA replication cycle. Without the DNA methyltransferase (DNMT), the replication machinery itself would produce daughter strands that are unmethylated and, over time, would lead to passive demethylation. DNMT1 is the proposed maintenance methyltransferase that is responsible for copying DNA methylation patterns to the daughter strands during DNA replication. Mouse models with both copies of DNMT1 deleted are embryonic lethal at approximately day 9, due to the requirement of DNMT1 activity for development in mammalian cells.
It is thought that DNMT3a and DNMT3b are the de novo methyltransferases that set up DNA methylation patterns early in development. DNMT3L is a protein that is homologous to the other DNMT3s but has no catalytic activity. Instead, DNMT3L assists the de novo methyltransferases by increasing their ability to bind to DNA and stimulating their activity. Finally, DNMT2 (TRDMT1) has been identified as a DNA methyltransferase homolog, containing all 10 sequence motifs common to all DNA methyltransferases; however, DNMT2 (TRDMT1) does not methylate DNA but instead methylates cytosine-38 in the anticodon loop of aspartic acid transfer RNA.
Since many tumor suppressor genes are silenced by DNA methylation during carcinogenesis, there have been attempts to re-express these genes by inhibiting the DNMTs. 5-Aza-2'-deoxycytidine (decitabine) is a nucleoside analog that inhibits DNMTs by trapping them in a covalent complex on DNA by preventing the β-elimination step of catalysis, thus resulting in the enzymes' degradation. However, for decitabine to be active, it must be incorporated into the genome of the cell, which can cause mutations in the daughter cells if the cell does not die. In addition, decitabine is toxic to the bone marrow, which limits the size of its therapeutic window. These pitfalls have led to the development of antisense RNA therapies that target the DNMTs by degrading their mRNAs and preventing their translation. However, it is currently unclear whether targeting DNMT1 alone is sufficient to reactivate tumor suppressor genes silenced by DNA methylation.
In plants
Significant progress has been made in understanding DNA methylation in the model plant Arabidopsis thaliana. DNA methylation in plants differs from that of mammals: while DNA methylation in mammals mainly occurs on the cytosine nucleotide in a CpG site, in plants the cytosine can be methylated at CpG, CpHpG, and CpHpH sites, where H represents any nucleotide but guanine. Overall, Arabidopsis DNA is highly methylated, mass spectrometry analysis estimated 14% of cytosines to be modified.
The principal Arabidopsis DNA methyltransferase enzymes, which transfer and covalently attach methyl groups onto DNA, are DRM2, MET1, and CMT3. Both the DRM2 and MET1 proteins share significant homology to the mammalian methyltransferases DNMT3 and DNMT1, respectively, whereas the CMT3 protein is unique to the plant kingdom. There are currently two classes of DNA methyltransferases: 1) the de novo class, or enzymes that create new methylation marks on the DNA; and 2) a maintenance class that recognizes the methylation marks on the parental strand of DNA and transfers new methylation to the daughters strands after DNA replication. DRM2 is the only enzyme that has been implicated as a de novo DNA methyltransferase. DRM2 has also been shown, along with MET1 and CMT3 to be involved in maintaining methylation marks through DNA replication. Other DNA methyltransferases are expressed in plants but have no known function (see the Chromatin Database).
It is not clear how the cell determines the locations of de novo DNA methylation, but evidence suggests that, for many (though not all) locations, RNA-directed DNA methylation (RdDM) is involved. In RdDM, specific RNA transcripts are produced from a genomic DNA template, and this RNA forms secondary structures called double-stranded RNA molecules. The double-stranded RNAs, through either the small interfering RNA (siRNA) or microRNA (miRNA) pathways direct de-novo DNA methylation of the original genomic location that produced the RNA. This sort of mechanism is thought to be important in cellular defense against RNA viruses and/or transposons, both of which often form a double-stranded RNA that can be mutagenic to the host genome. By methylating their genomic locations, through an as yet poorly understood mechanism, they are shut off and are no longer active in the cell, protecting the genome from their mutagenic effect.
In fungi

Many fungi have low levels (0.1 to 0.5%) of cytosine methylation, whereas other fungi have as much as 5% of the genome methylated. This value seems to vary both among species and among isolates of the same species. There is also evidence that DNA methylation may be involved in state-specific control of gene expression in fungi. However, at a detection limit of 250 attomoles by using ultra-high sensitive mass spectrometry DNA methylation was not confirmed in single cellular yeast species such as Saccharomyces cerevisiae or Schizosaccharomyces pombe, indicating that yeasts do not possess this DNA modification.
Although brewers' yeast (Saccharomyces) and fission yeast (Schizosaccharomyces) have no detectable DNA methylation, the model filamentous fungus Neurospora crassa has a well-characterized methylation system. Several genes control methylation in Neurospora and mutation of the DNA methyl transferase, dim-2, eliminates all DNA methylation but does not affect growth or sexual reproduction. While the Neurospora genome has very little repeated DNA, half of the methylation occurs in repeated DNA including transposon relics and centromeric DNA. The ability to evaluate other important phenomena in a DNA methylase-deficient genetic background makes Neurospora an important system in which to study DNA methylation.
In insects
DNA methylation is debated in insects, Drosophila melanogaster for instance seems to possess a very low level of DNA methylation only that is however too low to be studied by methods such as bisulphite sequencing. Takayama et al. developed a sensitive method that allowed to find that the fly genome DNA sequence patterns that associate with methylation are very different from the patterns seen in humans, or in other animal or plant species to date. Genome methylation in D. melanogaster was found at specific short motifs (concentrated in specific 5-base sequence motifs that are CA- and CT-rich but depleted of guanine) and is independent of DNMT2 activity.
In bacteria
Adenine or cytosine methylation is part of the restriction modification system of many bacteria, in which specific DNA sequences are methylated periodically throughout the genome. A methylase is the enzyme that recognizes a specific sequence and methylates one of the bases in or near that sequence. Foreign DNAs (which are not methylated in this manner) that are introduced into the cell are degraded by sequence-specific restriction enzymes and cleaved. Bacterial genomic DNA is not recognized by these restriction enzymes. The methylation of native DNA acts as a sort of primitive immune system, allowing the bacteria to protect themselves from infection by bacteriophage.
E. coli DNA adenine methyltransferase (Dam) is an enzyme of ~32 kDa that does not belong to a restriction/modification system. The target recognition sequence for E. coli Dam is GATC, as the methylation occurs at the N6 position of the adenine in this sequence (G meATC). The three base pairs flanking each side of this site also influence DNAâ€"Dam binding. Dam plays several key roles in bacterial processes, including mismatch repair, the timing of DNA replication, and gene expression. As a result of DNA replication, the status of GATC sites in the E. coli genome changes from fully methylated to hemimethylated. This is because adenine introduced into the new DNA strand is unmethylated. Re-methylation occurs within two to four seconds, during which time replication errors in the new strand are repaired. Methylation, or its absence, is the marker that allows the repair apparatus of the cell to differentiate between the template and nascent strands. It has been shown that altering Dam activity in bacteria results in increased spontaneous mutation rate. Bacterial viability is compromised in dam mutants that also lack certain other DNA repair enzymes, providing further evidence for the role of Dam in DNA repair.
One region of the DNA that keeps its hemimethylated status for longer is the origin of replication, which has an abundance of GATC sites. This is central to the bacterial mechanism for timing DNA replication. SeqA binds to the origin of replication, sequestering it and thus preventing methylation. Because hemimethylated origins of replication are inactive, this mechanism limits DNA replication to once per cell cycle.
Expression of certain genes, for example those coding for pilus expression in E. coli, is regulated by the methylation of GATC sites in the promoter region of the gene operon. The cells' environmental conditions just after DNA replication determine whether Dam is blocked from methylating a region proximal to or distal from the promoter region. Once the pattern of methylation has been created, the pilus gene transcription is locked in the on or off position until the DNA is again replicated. In E. coli, these pilus operons have important roles in virulence in urinary tract infections. It has been proposed that inhibitors of Dam may function as antibiotics.
On the other hand, DNA cytosine methylase targets CCAGG and CCTGG sites to methylate cytosine at the C5 position (C meC(A/T) GG). The other methylase enzyme, EcoKI, causes methylation of adenines in the sequences AAC(N6)GTGC and GCAC(N6)GTT.
Molecular cloning
Most strains used by molecular biologists are derivatives of E. coli K-12, and possess both Dam and Dcm, but there are commercially available strains that are dam-/dcm- (lack of activity of either methylase). In fact, it is possible to unmethylate the DNA extracted from dam+/dcm+ strains by transforming it into dam-/dcm- strains. This would help digest sequences that are not being recognized by methylation-sensitive restriction enzymes.
The Restriction enzyme DpnI can recognize 5'-GmeATC-3'sites and digest the methylated DNA. Being such a short motif, it occurs frequently in sequences by chance, and as such its primary use for researchers is to degrade template DNA following PCR reactions (PCR products lack methylation, as no methylases are present in the reaction). Similarly, some commercially available restriction enzymes are sensitive to methylation at their cognate restriction sites, and must as mentioned previously be used on DNA passed through a dam-/dcm- strain to allow cutting.
Detection
DNA methylation can be detected by the following assays currently used in scientific research:
- Mass spectrometry is a very sensitive and reliable analytical method to detect DNA methylation. MS in general is however not informative about the sequence context of the methylation, thus limited in studying the function of this DNA modification.
- Methylation-Specific PCR (MSP), which is based on a chemical reaction of sodium bisulfite with DNA that converts unmethylated cytosines of CpG dinucleotides to uracil or UpG, followed by traditional PCR. However, methylated cytosines will not be converted in this process, and primers are designed to overlap the CpG site of interest, which allows one to determine methylation status as methylated or unmethylated.
- Whole genome bisulfite sequencing, also known as BS-Seq, which is a high-throughput genome-wide analysis of DNA methylation. It is based on aforementioned sodium bisulfite conversion of genomic DNA, which is then sequenced on a Next-generation sequencing platform. The sequences obtained are then re-aligned to the reference genome to determine methylation states of CpG dinucleotides based on mismatches resulting from the conversion of unmethylated cytosines into uracil.
- The HELP assay, which is based on restriction enzymes' differential ability to recognize and cleave methylated and unmethylated CpG DNA sites.
- ChIP-on-chip assays, which is based on the ability of commercially prepared antibodies to bind to DNA methylation-associated proteins like MeCP2.
- Restriction landmark genomic scanning, a complicated and now rarely used assay based upon restriction enzymes' differential recognition of methylated and unmethylated CpG sites; the assay is similar in concept to the HELP assay.
- Methylated DNA immunoprecipitation (MeDIP), analogous to chromatin immunoprecipitation, immunoprecipitation is used to isolate methylated DNA fragments for input into DNA detection methods such as DNA microarrays (MeDIP-chip) or DNA sequencing (MeDIP-seq).
- Pyrosequencing of bisulfite treated DNA. This is sequencing of an amplicon made by a normal forward primer but a biotinylated reverse primer to PCR the gene of choice. The Pyrosequencer then analyses the sample by denaturing the DNA and adding one nucleotide at a time to the mix according to a sequence given by the user. If there is a mis-match, it is recorded and the percentage of DNA for which the mis-match is present is noted. This gives the user a percentage methylation per CpG island.
- Molecular break light assay for DNA adenine methyltransferase activity â€" an assay that relies on the specificity of the restriction enzyme DpnI for fully methylated (adenine methylation) GATC sites in an oligonucleotide labeled with a fluorophore and quencher. The adenine methyltransferase methylates the oligonucleotide making it a substrate for DpnI. Cutting of the oligonucleotide by DpnI gives rise to a fluorescence increase.
- Methyl Sensitive Southern Blotting is similar to the HELP assay, although uses Southern blotting techniques to probe gene-specific differences in methylation using restriction digests. This technique is used to evaluate local methylation near the binding site for the probe.
- MethylCpG Binding Proteins (MBPs) and fusion proteins containing just the Methyl Binding Domain (MBD) are used to separate native DNA into methylated and unmethylated fractions. The percentage methylation of individual CpG islands can be determined by quantifying the amount of the target in each fraction. Extremely sensitive detection can be achieved in FFPE tissues with abscription-based detection.
- High Resolution Melt Analysis (HRM or HRMA), is a post-PCR analytical technique. The target DNA is treated with sodium bisulfite, which chemically converts unmethylated cytosines into uracils, while methylated cytosines are preserved. PCR amplification is then carried out with primers designed to amplify both methylated and unmethylated templates. After this amplification, highly methylated DNA sequences contain a higher number of CpG sites compared to unmethylated templates, which results in a different melting temperature that can be used in quantitative methylation detection.
Differentially methylated regions (DMRs)
Differentially methylated regions (DMRs), are genomic regions with different methylation statuses among multiple samples (tissues, cells, individuals or others), are regarded as possible functional regions involved in gene transcriptional regulation. The identification of DMRs among multiple tissues (T-DMRs) provides a comprehensive survey of epigenetic differences among human tissues. DMRs between cancer and normal samples (C-DMRs) demonstrate the aberrant methylation in cancers. It is well known that DNA methylation is associated with cell differentiation and proliferation. Many DMRs have been found in the development stages (D-DMRs) and in the reprogrammed progress (R-DMRs). In addition, there are intra-individual DMRs (Intra-DMRs) with longitudinal changes in global DNA methylation along with the increase of age in a given individual. There are also inter-individual DMRs (Inter-DMRs) with different methylation patterns among multiple individuals.
QDMR (Quantitative Differentially Methylated Regions) is a quantitative approach to quantify methylation difference and identify DMRs from genome-wide methylation profiles by adapting Shannon entropy (http://bioinfo.hrbmu.edu.cn/qdmr). The platform-free and species-free nature of QDMR makes it potentially applicable to various methylation data. This approach provides an effective tool for the high-throughput identification of the functional regions involved in epigenetic regulation. QDMR can be used as an effective tool for the quantification of methylation difference and identification of DMRs across multiple samples.
Gene-set analysis (a.k.a pathway analysis; usually performed tools such as DAVID, GoSeq or GSEA) has been shown to be severely biased when applied to high-throughput methylation data (e.g. MeDIP-seq, MeDIP-ChIP, HELP-seq etc.), and a wide range of studies have thus mistakenly reported hyper-methylation of genes related to development and differentiation; it has been suggested that this can be corrected using sample label permutations or using a statistical model to control for differences in the numberes of CpG probes / CpG sites that target each gene.
Computational prediction

DNA methylation can also be detected by computational models through sophisticated algorithms and methods. Computational models can facilitate the global profiling of DNA methylation across chromosomes, and often such models are faster and cheaper to perform than biological assays. Such up-to-date computational models include Bhasin, et al., Bock, et al., and Zheng, et al. Together with biological assay, these methods greatly facilitate the DNA methylation analysis.





FINANCIAL BREAKTHROUGH
ReplyDeleteLife indeed is GRACE, I'am Daan Sophia currently in California USA. I would like to share my experience with you guys on how I got a loan of $185,000.00 USD to clear my bank draft and start up a new business. It all started when i lost my home and belongings due to the bank draft I took to offset some bills and some personal needs. I became so desperate and began to seek for funds at all means. Luckily for me I heard a colleague of mine talking about this company, I got interested although i was scared of being scammed, I was compelled by my situation and had no choice than to seek advise from my friend regarding this very company and was given their contact number, getting intouch with them really made me skeptical due to my past experience with online lenders, little did i know this very Company "PROGRESSIVE LOAN INC. was a godsent to me and my family and the entire Internet World, this company has been of great help to me and some of my colleague and today am a proud owner of well organized business and responsibilities are well handled all thanks to Josef Lewis of (progresiveloan@yahoo.com).. So if really you are genuinely in need of a loan either to expand or start up your own business or in any form of financial difficulty, i advise you give Mr Josef Lewis of Progressive loan the opportunity of financial upliftment in your life Email: progresiveloan@yahoo.com OR Call/Text +1(603) 786-7565 and not fall victim of online scam in the name of getting a loan. thanks
Some days, things do not happen the way you want them to be. They fall
Deleteapart, and you start to worry. Worse, you feel discouraged and lonely,
thinking that there is no other way to straighten things up Natural herbs
have cured so many illness that drugs and injection can't cure I've seen
the great importance of natural herbs and the wonderful work natural herbs
have done in people's lives. i read a lot of people's testimonies online on
how they were cured of Herpes, HIV, Diabetics, Lymphoma, Fibroid, Cancer
etc by Dr voodoo herbal medicine, so i decided to contact the doctor Dr
voodoo also cured me from HIV I will recommend Dr. voodoo to everyone
reading this article because this herbal healer is capable to heal anything
email Dr voodoo at: voodoospelltemple66@gmail.com also his whatsApp
+2348140120719
This could be reason behind ghastly outcome for understudy calling and for individuals. Our http://pathology.residencypersonalstatements.net/ this issue from understudy business.
ReplyDeletePeople who have to face serious obstacles sometimes develop remarkable adaptations that make them rather unique individuals.
ReplyDeleteDrug rehab center Indianapolis
I found this information very useful.
ReplyDeleteIt was during my research on HIV/Herpes that I stumbled upon the Hiv/Herpes information; information which is quite easy to find when doing a search for STD on google. I was into conspiracy at the time thought of HIV/Herpes Cured' being a conspiracy was something Ignorance though,I found pretty interesting about herbal medicine. I asked questions about the Herbal cure's on official HIV/Herpes websites and I was banned for doing so by moderators who told me that I was parroting Hiv/Herpes propaganda. This reinforced my belief that there is a cure for Hiv/Herpes Then i found a lady from germany name Achima Abelard Dr Itua Cure her Hiv so I send him a mail about my situation then talk more about it and send me his herbal medicine I drank for two weeks.And today I'm Cured no Hiv/Herpes in my life,I searched for Hiv/Herpes groups to attempt to make contact with people in order to learn more about Hiv/Herpes Herbal Cure's I believed at this time that you with the same disease this information is helpful to you and I wanted to do the best I could to spread this information in the hopes of helping other people.That Dr Itua Herbal Medicine makes me believes there is a hope for people suffering from,Parkinson's disease,Schizophrenia,Cancer,Scoliosis,Fibromyalgia,Fluoroquinolone Toxicity Syndrome Fibrodysplasia Ossificans Progressiva.Fatal Familial Insomnia Factor V Leiden Mutation ,Epilepsy Dupuytren's disease,Desmoplastic small-round-cell tumor Diabetes ,Coeliac disease,Creutzfeldt–Jakob disease,Cerebral Amyloid Angiopathy, Ataxia,Arthritis,Amyotrophic Lateral Sclerosis,Alzheimer's disease,Adrenocortical carcinoma.Asthma,Allergic diseases.Hiv_ Aids,Herpes,Inflammatory bowel disease ,Copd,Diabetes,Hepatitis,I read about him online how he cure Tasha and Tara,Conley,Mckinney and many more suffrin from all kind of disease so i contacted him . He's a herbal doctor with a unique heart of God, Contact Emal..drituaherbalcenter@gmail.com Phone or whatsapp..+2348149277967.
ReplyDelete6 months ago I was diagnosed with COPD with 55-60% lung capacity. My doctor just said all the crying , stomping your feet will not change it so just accept it and basically patted me on the back and sent me home to die. I was devastated and was afraid to do anything. I stopped riding my bike, I was afraid to do anything that would cause any exertion. It consumed my thoughts with every breath and the fear of what to expect was almost more than I could deal with. So I couldn't get myself all time I decided to find a herbal cure online and I came acrose Doctor Itua on how he cure several people suffering from Hiv, and Herpes I gave him a call on this Number +2348149277967 also chat on whatsapp he gave me all the details about the cure i paid him for the medicine after 5 working days i receieve my herbal medicine ,I use it for two weeks that is how I get Cured and today I'm living healthy and fine I give him thanks also promise him to testify about his work,He Can also cure the following deseases..Copd,Herpes,Alzheimer's disease,Adrenocortical carcinoma.Asthma,Allergic diseases.Parkinson's Fatal Familial Insomnia Factor V Leiden Mutation ,Epilepsy Dupuytren's disease,disease,Schizophrenia,Cancer,Scoliosis,Fibromyalgia,Hiv,Hepatitis,diabetes,Coeliac disease,Creutzfeldt–Jakob disease,Cerebral Amyloid Angiopathy, Asthma.Contact...drituaherbalcenter@gmail.com..Whatsapp Number...+2348149277976
ReplyDeleteDr Itua cure my HIV, I have been a ARV Consumption for 10 years. i have been in pains until i came across Dr Itua on blogs site.I emailed him about my details of my HIV and my location i explained every thing to him and he told me that there is nothing to be scared of that he will cured me, he gave me guarantee,He ask me to pay for items fees so when i'm cured I will show gratitude I did and giving testimony of his healing herbs is what I'm going to do for the rest of you out there having HIV and other disease can see the good work of Dr Itua.I received his herbal medicine through EMS Courier service who delivered to my post office within 5 working days.Dr Itua is an honest man and I appreciate him for his good work.My GrandMa called him to appreciate him and rest of my friends did too,Is a joy to me that I'm free of taking Pills and having that fat belle is a nightmare.you will understand what i'm talking about if you have same problem I was having then not now though.I'm free and healthy Big Thanks To Dr Itua Herbal Center.I have his calendar too that he recently sent me,He Cure all kind disease Like,Cancer,Fibromyalgia,Fluoroquinolone Toxicity
ReplyDeleteSyndrome Fibrodysplasia Ossificans Progressiva.,Herpes,Hiv,Hepatitis B,Fibroid,Diabetes,Dercum,Alzheimer's disease,Adrenocortical carcinoma.Asthma,Allergic diseases.Copd ,and also Bring back Ex Lover Back..Here his Contact .drituaherbalcenter@gmail.com Or Whats_app Number +2348149277967
Irrespective of receiving daily oral or future injectable depot therapies, these require health care visits for medication and monitoring of safety and response. If patients are treated early enough, before a lot of immune system damage has occurred, life expectancy is close to normal, as long as they remain on successful treatment. However, when patients stop therapy, virus rebounds to high levels in most patients, sometimes associated with severe illness because i have gone through this and even an increased risk of death. The aim of “cure”is ongoing but i still do believe my government made millions of ARV drugs instead of finding a cure. for ongoing therapy and monitoring. ARV alone cannot cure HIV as among the cells that are infected are very long-living CD4 memory cells and possibly other cells that act as long-term reservoirs. HIV can hide in these cells without being detected by the body’s immune system. Therefore even when ART completely blocks subsequent rounds of infection of cells, reservoirs that have been infected before therapy initiation persist and from these reservoirs HIV rebounds if therapy is stopped. “Cure” could either mean an eradication cure, which means to completely rid the body of reservoir virus or a functional HIV cure, where HIV may remain in reservoir cells but rebound to high levels is prevented after therapy interruption.Dr Itua Herbal Medicine makes me believes there is a hope for people suffering from,Parkinson's disease,Schizophrenia,Cancer,Scoliosis,Fibromyalgia,Fluoroquinolone Toxicity
ReplyDeleteSyndrome Fibrodysplasia Ossificans Progressiva.Fatal Familial Insomnia Factor V Leiden Mutation ,Epilepsy Dupuytren's disease,Desmoplastic small-round-cell tumor Diabetes ,Coeliac disease,Creutzfeldt–Jakob disease,Cerebral Amyloid Angiopathy, Ataxia,Arthritis,Amyotrophic Lateral Sclerosis,Alzheimer's disease,Adrenocortical carcinoma.Asthma,Allergic diseases.Hiv_ Aids,Herpes,Inflammatory bowel disease ,Copd,Diabetes,Hepatitis,I read about him online how he cure Tasha and Tara so i contacted him on drituaherbalcenter@gmail.com even talked on whatsapps +2348149277967 believe me it was easy i drank his herbal medicine for two weeks and i was cured just like that isn't Dr Itua a wonder man? Yes he is! I thank him so much so i will advise if you are suffering from one of those diseases Pls do contact him he's a nice man.
My life is beautiful thanks to you, Mein Helfer. Lord Jesus in my life as a candle light in the darkness. You showed me the meaning of faith with your words. I know that even when I cried all day thinking about how to recover, you were not sleeping, you were dear to me. I contacted the herbal center Dr Itua, who lived in West Africa. A friend of mine here in Hamburg is also from Africa. She told me about African herbs but I was nervous. I am very afraid when it comes to Africa because I heard many terrible things about them because of my Christianity. god for direction, take a bold step and get in touch with him in the email and then move to WhatsApp, he asked me if I can come for treatment or I want a delivery, I told him I wanted to know him I buy ticket in 2 ways to Africa To meet Dr. Itua, I went there and I was speechless from the people I saw there. Patent, sick people. Itua is a god sent to the world, I told my pastor about what I am doing, Pastor Bill Scheer. We have a real battle beautifully with Spirit and Flesh. Adoration that same night. He prayed for me and asked me to lead. I spent 2 weeks and 2 days in Africa at Dr Itua Herbal Home. After the treatment, he asked me to meet his nurse for the HIV test when I did it. It was negative, I asked my friend to take me to another nearby hospital when I arrived, it was negative. I was overwhite with the result, but happy inside of me. We went with Dr. Itua, I thank him but I explain that I do not have enough to show him my appreciation, that he understands my situation, but I promise that he will testify about his good work. Thank God for my dear friend, Emma, I know I could be reading this now, I want to thank you. And many thanks to Dr. Itua Herbal Center. He gave me his calendar that I put on my wall in my house. Dr. Itua can also cure the following diseases ... Cancer, HIV, Herpes, Hepatitis B, Inflammatory Liver, Diabetis, Fribroid,Parkinson's disease,Inflammatory bowel disease ,Fibromyalgia, recover your ex. You can contact him by email or whatsapp, @ .. drituaherbalcenter@gmail.com, phone number .. + 2348149277967 .. He is a good doctor, talk to him kindly. I'm sure he will also listen to you.
ReplyDeleteI'm 55-year-old from Korean, I was diagnosed with second-stage liver cancer following a scheduled examination to monitor liver cirrhosis. I had lost a lot of weight. A CT scan revealed three tumors; one in the center of my liver in damaged tissue and two in healthy portions of my liver. No chemotherapy or radiotherapy treatment was prescribed due to my age, the number of liver tumors. One month following my diagnosis I began taking 12 (350 point) Salvestrol supplements per day, commensurate with my body weight. This comprised six Salvestrol Shield (350 point) capsules and six Salvestrol Gold (350 point) capsules, spread through the day by taking two of each capsule after each main meal. This level of Salvestrol supplementation (4,000 points per day) was maintained for four months. In addition, I began a program of breathing exercises, chi exercises, meditation, stretching and stress avoidance. Due to the variety of conditions that I suffered from, I received ongoing medical examinations. Eleven months after commencing Salvestrol supplementation But all invalid so I keep searching for a herbal cure online that how I came across a testimony appreciating Dr Itua on how he cured her HIV/Herpes, I contacted him through email he listed above, Dr Itua sent me his herbal medicine for cancer to drink for two weeks to cure I paid him for the delivering then I received my herbal medicine and drank it for two weeks and I was cured until now I'm all clear of cancer, I will advise you to contact Dr Itua Herbal Center On Email...drituaherbalcenter@gmail.com. WhatsApps Number...+2348149277967. If you are suffering from Diseases listed below, Cancer, HIV/Aids, Herpes Virus, Hepatitis, Chronic Illness. Lupus,Fibromyalgia.
ReplyDeleteGod bless Dr Larry for his marvelous work in my life, I was diagnosed of HERPES SIMPLEX VIRUS since 2010 and I was taking my medications, I wasn't satisfied i needed to get the HERPES out of my system, I searched about some possible cure for HERPES i saw a comment about Dr Larry, how he cured HERPES with his herbal medicine, I contacted him and he guided me. I asked for solutions, he started the remedy for my health, he sent me the medicine within 3days. I took the medicine as prescribed by him and 2weeks later i was cured from HERPES contact him assurancesolutionhome@gmail.com once again thanks to you Dr Larry. cure the flowing virus, contact his web site: http://assurancesolutionhome.wordpress.com
ReplyDeletehttp://assurancesolutionhome.website2.me/ or add him on whatsapp +1(424)-261-8520
cancer cure
diabetes cure
ringing ear
herpes cure
warts cure
HPV cure
HIV
get your ex back
pregnancy herbal medicine
prostate enlargement
Hepatitis
God bless Dr Larry for his marvelous work in my life, I was diagnosed of HERPES SIMPLEX VIRUS since 2010 and I was taking my medications, I wasn't satisfied i needed to get the HERPES out of my system, I searched about some possible cure for HERPES i saw a comment about Dr Larry, how he cured HERPES with his herbal medicine, I contacted him and he guided me. I asked for solutions, he started the remedy for my health, he sent me the medicine within 3days. I took the medicine as prescribed by him and 2weeks later i was cured from HERPES contact him assurancesolutionhome@gmail.com once again thanks to you Dr Larry. cure the flowing virus, contact his web site: http://assurancesolutionhome.wordpress.com
http://assurancesolutionhome.website2.me/ or add him on whatsapp +1(424)-261-8520
cancer cure
diabetes cure
ringing ear
herpes cure
warts cure
HPV cure
HIV
get your ex back
pregnancy herbal medicine
prostate enlargement
Hepatitis
ReplyDeleteI am bold enough among many others to state that there is now a potent cure to this sickness but many are unaware of it. I discovered that I was infected with the virus 3 months ago, after a medical check-up. My doctor told me and I was shocked, confused and felt like my world has crumbled. I was dying slowly due to the announcement of my medical practitioner but he assured me that I could leave a normal life if I took my medications (as there was no medically known cure to Herpes). I went from churches to churches but soon found that my case needed urgent attention as I was growing lean due to fear of dying anytime soon. In a bid to look for a lasting solution to my predicament, I sought for solutions from the herbal world. I went online and searched for every powerful trado-medical practitioner that I could severe, cos I heard that the African Herbs had a cure to the Herpes syndrome. It was after a little time searching the web that I came across one Dr Itua(A powerful African Herbal Doctor), who offered to help me at a monetary fee. I had to comply as this was my final bus-stop to receiving a perfect healing. My last resolve was to take my life by myself, should this plan fail. At last it worked out well. He gave me some steps to follow and I meticulously carried out all his instructions. Last month, to be precise, I went back to the hospital to conduct another test and to my amazement, the results showed that negative,Dr Itua Can As Well Cure The Following Desease…Cancer,Hiv,Herpes, Hepatitis B,Liver Inflammatory,Diabetis,Fribroid,,Non Hodgkin Lymphoma,Skin Cancer,Uterine Cancer,Prostate Cancer Dercum,Infertility,fibromyalgia,Get Your Ex Back,Als,SYPHILLIS,Genetic disease,Epilepsy, Parkinson's disease..You can free yourself of this Herpes virus by consulting this great African Herbal Doctor via this e-mail: drituaherbalcenter@gmail.com or call and whatsapp him on +2348149277967 He will help you and his herb medication is sure. he has the cure on all disease .You can talk to me on INSTAGRAM..tashamoore219....
Although therapy has become more tolerable and once daily FDC tables have made it easier to adhere to prescribed treatment, it still requires that patients take their medication regularly to achieve sustained viral suppression. When treatment adherence is inadequate and replication is therefore not suppressed, But dr itua promised and fulfilled his promised to me as he said I will share his work to people that are suffering from Infertility, Herpes, Hepatitis A/B, Fibroid, HIV/ Aids, Alzheimer's disease, Arthritis, Copd, Diabetes, Liver/Kidney Inflamotry, Fibromyalgia, Parkinson's disease, I have read a lot of testimony online from Jesus McKinney,Achima Abelard and Tara Omar on how dr itua heal them with his herbal medicine I contacted him on Email drituaherbalcenter@gmail.com then we talk on whatsapp +2348149277967 he gave me instruction on how to drink it for two weeks then after drinking it for two weeks I went for test then I find out I was cured of HIV, I thank him allot i also send him some money for appreciation, Contact this great herbal doctor if you are a sick person.
ReplyDeleteMost prostate cancers are adenocarcinomas, cancers that arise in glandular cells of the prostate’s epithelial tissue. Prostate cancers usually progress slowly and produce no symptoms in the initial stages. Eventually, the tumor may enlarge like mine use too, the prostate gland, pressing on the urethra and causing painful or frequent urination and blood in the urine. So I was so uncomfortable with this prostate cancer diseases then I decided to do online search on how to cure cancer because I well have read a lot about herbal medicine,I came across a lot of testimony how Dr Itua cure HIV/herpes then Cancer was listed below the comment.with courage I contacted Dr Itua and he sent me his herbal medicine through Courier service then I was asked to pick it up at my post office which i quickly did. I contacted Dr Itua that i have received my herbal medicine so he instructs me on how to drink it for three weeks and that is how Dr Itua Herbal Medicine cure my prostate Cancer, The treatment takes three weeks and I was cured completely. Dr Itua is a god sent and I thank him every day of my life. Contact him now On:Email:drituaherbalcenter@gmail.com/Whatsapp:+2348149277967.
ReplyDeleteHe listed to that he can as well cure the following diseases below.... Cerebral Amides. Lung Cancer, Alzheimer's disease, Adrenocortical carcinoma. Alma, Uterine Cancer, Breast Cancer, Allergic diseases. Kidney cancer, Love Spell, Colo rectal cancer, Lottery Spell, Bladder Cancer, Skin Cancer, HIV /Aids, Herpes, Non-Hodgkin lymphoma, Inflammatory bowel disease, Copd, Diabetes, Hepatitis
Hydrocodone side effects
ReplyDeleteGet emergency medical help if you have signs of an allergic reaction to hydrocodone: hives; difficult breathing swelling of your face, lips, tongue, or throat.
Stop usinghydrocodone and call your doctor at once if you have:
noisy breathing, sighing, shallow breathing;
a slow heart rate or weak pulse;
pain or burning when you urinate;
confusion, tremors, severe drowsiness;
a light-headed feeling, like you might pass out; or
low cortisol levels - nausea, vomiting, loss of appetite, dizziness, worsening tiredness or weakness.
I'm 61 years old, I contracted hpv in 2011' I has be taking lot treatment for it and some months ago the wart stated coming out seriously, I used lot recommendation because there was lot warts around my anus and was so embarrassed. but today I'm totally happy I got the virus eliminated by using natural treatment from Dr Onokun herbal center after his treatment I got cured. all the warts went away' seriously believed Dr Onokun he have the cure for human papillomavirus because he has eliminated hpv been in my body since 2011, Dr Onokun make it possible for me. Here is Dr Onokun email to reach him: Dronokunherbalcure@gmail.com he is welled capable of curing terrible diseases.
ReplyDeleteJoy and happiness is all i see around ever since i came in contact with this great man. i complained bitterly to him about me having herpes only for him to tell me it’s a minor stuff. He told me he has cured thousands of people but i did not believe until he sent me the herbal medicine and i took it as instructed by this great man, only to go to the hospital after two weeks for another test and i was confirmed negative. For the first time in four years i was getting that result. i want to use this medium to thank this great man. His name is Dr aziegbe, i came in contact with his email through a friend in UK and ever since then my live has been full with laughter and great peace of mind. i urge you all with herpes or HSV to contact him if you willing to give him a chance. you can contact him through this email DRAZIEGBE1SPELLHOME@GMAIL .COM or you can also WhatsApp him +2349035465208
ReplyDeleteHe also cured my friend with HIV and ever since then i strongly believe he can do all things. Don't be deceived thinking he does not work, i believe if you can get in contact with this man all your troubles will be over. i have done my part in spreading the good news. Contact him through his email and you will be the next to testify of his great work.
Happiness is all i see now I never thought that I will live on
ReplyDeleteearth before the year runs out. I have been suffering from a
deadly disease (Herpes) for the past 3 years now; I had spent
a lot of money going from one places to another, from
churches to churches, hospitals have been my home every day
residence. Constant checks up have been my hobby not until
this faithful day, I was searching through the internet, I saw a
testimony on how pp him +2348154637647 Dr Lucky, helped
someone in curing his Herpes disease, quickly I copied his
email which is (drluckyherbalcure@gmail.com) just to give
him a test I spoke to him, he asked me to do some certain
things which I did, he told me that he is going to provide the
herbal cure to me, which he did, then he asked me to go for
medical checkup after some days after using the herbal cure, I
was free from the deadly disease, he only asked me to post
the testimony through the whole world, faithfully am doing it
now, please brothers and sisters, he is great, I owe him in
return. if you are having a similar problem just email him on
(drluckyherbalcure@gmail.com) or Call him or WhatsApp him
+2348154637647
Do this hack to drop 2lb of fat in 8 hours
ReplyDeleteOver 160000 women and men are using a easy and secret "liquids hack" to lose 1-2 lbs each and every night while they sleep.
It is easy and works every time.
This is how to do it yourself:
1) Take a glass and fill it up half glass
2) Then use this awesome hack
so you'll become 1-2 lbs skinnier as soon as tomorrow!
ReplyDeletei couldn't believe that i would ever be re-unite with my ex-lover, i was so traumatize staying all alone with no body to stay by me and to be with me, but i was so lucky one certain day to meet this powerful spell caster Dr Akhere,after telling him about my situation he did everything humanly possible to see that my lover come back to me,indeed after casting the spell my ex-lover came back to me less than 48 hours,my ex-lover came back begging me that he will never leave me again,3 months later we got engaged and married,if you are having this same situation just contact Dr Akhere on his email: AKHERETEMPLE@gmail.com thanks very much sir for restoring my ex-lover back to me,his email: AKHERETEMPLE@gmail.com or call/whatsapp:+2349057261346
hindi ako makapaniwala na kailanman ay muling makiisa ako sa aking kasintahan, labis akong na-trauma sa pananatiling nag-iisa na walang katawan na manatili sa akin at makakasama ko, ngunit napakasuwerte ako sa isang tiyak na araw upang matugunan ito malakas na spell caster na si Dr Akhere, matapos sabihin sa kanya ang tungkol sa aking sitwasyon ginawa niya ang lahat ng makataong posible upang makita na ang aking kasintahan ay bumalik sa akin, sa katunayan matapos na ihagis ang spell ang aking dating kasintahan ay bumalik sa akin ng mas mababa sa 48 oras, dumating ang dating kasintahan ko. bumalik sa pagmamakaawa sa akin na hindi na niya ako pababayaan, 3 buwan mamaya kami ay nakipag-ugnay at nag-asawa, kung nagkakaroon ka ng parehong sitwasyong ito makipag-ugnay lamang kay Dr Akhere sa kanyang email: AKHERETEMPLE@gmail.com maraming salamat sa sir sa pagpapanumbalik ng aking dating kasintahan bumalik sa akin, ang kanyang email: AKHERETEMPLE@gmail.com o tumawag / whatsapp: +2349057261346
God bless Dr. LUCKY for his marvelous work in my life, I was diagnosed of HERPES since 2017 and I was taking my medications, I wasn’t satisfied i needed to get the HERPES out of my system, I searched out some possible cure for HERPES i saw a comment about Dr. LUCKY, how he cured HERPES,DIABETES,HIV,EX_LOVER BACK and CANCER with his herbal medicine, I contacted him and he guided me. I asked for solutions, he started the remedy for my health, he sent me the medicine I took the medicine as prescribed by him and 14 days later i was cured from HERPES, Dr. LUCKY truly you are great, do you need his help also? Why don’t you contact him through his EMAIL: (drluckyherbalcure@gmail.com ) Caal or whatsApp him on +2348154637647
ReplyDeleteHappiness is all i see now I never thought that I will live on
ReplyDeleteearth before the year runs out. I have been suffering from a
deadly disease (Herpes) for the past 3 years now; I had spent
a lot of money going from one places to another, from
churches to churches, hospitals have been my home every day
residence. Constant checks up have been my hobby not until
this faithful day, I was searching through the internet, I saw a
testimony on how pp him +2348154637647 Dr Lucky, helped
someone in curing his Herpes disease, quickly I copied his
email which is (drluckyherbalcure@gmail.com) just to give
him a test I spoke to him, he asked me to do some certain
things which I did, he told me that he is going to provide the
herbal cure to me, which he did, then he asked me to go for
medical checkup after some days after using the herbal cure, I
was free from the deadly disease, he only asked me to post
the testimony through the whole world, faithfully am doing it
now, please brothers and sisters, he is great, I owe him in
return. if you are having a similar problem just email him on
(drluckyherbalcure@gmail.com) or Call him or WhatsApp him
+2348154637647
I’m here to testify about what DR. ISIBOR did for me. I have been suffering from (GENITAL HERPES VIRUS) disease for the past 3 years and had constant pain and inching, especially in my private part. During the first year, I had faith in God that i would be cured someday.This disease started circulating all over my body and I have been taking treatment from my doctor, few weeks ago I came across a testimony of Rose Smith on the internet testifying about a Man called DR. ISIBOR on how he cured her from 7 years HSV 2. And she also gave the email address of this man, advise anybody to contact him for help on any kind of diseases that he would be of help, so I emailed him telling him about my (HSV 2) he told me not to worry that I was going to be cured!! Well, I never doubted him I have faith he can cure me too,, DR. ISIBOR prepared and sent me Healing Oil, Soap, roots and herbs which I took. In the first one week, I started experiencing changes all over me, after four weeks of using his Roots/ Herbs, Oil and Soap, I was totally cured. no more inching , pain on me anymore as DR. ISIBOR assured me. After some time I went to my doctor to do another test behold the result came out negative. So friends my advise is if you have such disease or know anyone who suffers from it or any other disease like HPV, HIV, ALS, CANCER etc. you can contact DR. ISIBOR for help via email} drisiborspellhome@gmail.com or call +2348107855231
ReplyDeleteCan't still believe that i got cured from Genital Herpes through herbal treatment from Dr LUCKY who I met through the internet, I actually couldn't believe it at first because it sounded impossible to me knowing how far I have gone just to get rid of it. Dr LUCKY send me his medicine which I took as instructed and here I am living a happy life once again, a big thanks to Dr LUCKY , I am sure there are many herbal doctors out there but Dr LUCKY did it for me, contact him on Email him; { drluckyherbalcure@gmail.com }
ReplyDelete
ReplyDeleteThank you Dr Williams for what you have done for me am so greatful my lover is back to me and we are now living happily together. Dr Williams love spell is very powerful and effective and it does not have any side effect as he promised I decided to give Dr Williams a try when my lover left me for another he helped me to cast a love spell on my lover that brought him back to me what makes me excited the most is that my lover did not even know he is under a spell if you are passing through relationship love problem I advice you to contact Dr Williams to get your problem solve. It's very hard to loose a love one and I know how it feels so do not let somebody take away your lover from you. Contact Dr Williams today and get your problem solve and if you need his help below here is his email address.
drwilliams533@gmail. com or you can also reach him on his WhatsApp +2348136785562
Are you paying $189 a week for Valtrex or $67 for Acyclovir to suppress your outbreaks? Have you considered your yearly prescription costs for these drugs, $2000+ a year! Anyone who has herpes can attest to the painful and embarrassing symptoms. Unfortunately, many find that doctor recommended products are frequently not effective at taking care of the problem.DR OHIKHOBO herbal remedy will get to the root of the cause and cure you completely rather than suppress outbreaks with medication. and leave you happier, healthier, and outbreak FREE!.let my tell you all how I was cured completely from herpes at first I never believed I could be cure considering all type of medication I have been using but it all came like dream come true for me that i was able to be cured by OHIKHOBO herbal remedy as I speak now I have no more outbreaks,.It could change your life too.contact DR OHIKHOBO at drohikhoboherbalcenter@gmail.com or WhatsApp +2348103601042
ReplyDeleteI’m here to testify about what DR. ISIBOR did for me. I have been suffering from (GENITAL HERPES VIRUS) disease for the past 3 years and had constant pain and inching, especially in my private part. During the first year, I had faith in God that i would be cured someday.This disease started circulating all over my body and I have been taking treatment from my doctor, few weeks ago I came across a testimony of Rose Smith on the internet testifying about a Man called DR. ISIBOR on how he cured her from 7 years HSV 2. And she also gave the email address of this man, advise anybody to contact him for help on any kind of diseases that he would be of help, so I emailed him telling him about my (HSV 2) he told me not to worry that I was going to be cured!! Well, I never doubted him I have faith he can cure me too,, DR. ISIBOR prepared and sent me Healing Oil, Soap, roots and herbs which I took. In the first one week, I started experiencing changes all over me, after four weeks of using his Roots/ Herbs, Oil and Soap, I was totally cured. no more inching , pain on me anymore as DR. ISIBOR assured me. After some time I went to my doctor to do another test behold the result came out negative. So friends my advise is if you have such disease or know anyone who suffers from it or any other disease like HPV, HIV, ALS, CANCER etc. you can contact DR. ISIBOR for help via email} {drisiborspellhome@gmail.com} you can also contact him on WhatsApp +2348107855231
ReplyDeleteI want to give a testimony on how I got cured from Herpes Virus. Few months back I was having some symptoms . I went to see a doctor and many blood tests was done on me, later on I was told I had Herpes. My doctor told me that there's no cure for Herpes I felt bad, I went online searching for a possible cure for Herpes Virus, I saw a post of a Herbal doctor that cured someone from Herpes. I contacted him and told him how I'm feeling he said his herbal medicine can cure me. He sent me the medicine via UPS Service and I received the medicine some days after he sent it, i took the medicine as prescribed by him. Before the completion of the medication the symptoms stopped. I went to the doctor and carried out another blood test, surprisingly I was Negative. I haven't had any symptoms anymore, you can contact him viaEmail drafridherbalhome@gmail.com or whatsapp number +2349057260738.
ReplyDeleteHerpes is a sexually transmitted virus that primarily infects the mouth and the genitals. It is transmitted by bodily fluids – penetration isn’t required for transmission, oral-oral or oral-genital contact will suffice.I WAS ONCE HERPES SIMPLEX VIRUS patient, I have HSV-I genitally and I'm one of the rare cases where I was infected by sexual contact. The person who infected me was unaware that he had it, and he tested negative on a blood test (he probably got it from his mother at birth and never had an outbreak in his life).I'm sure I have HSV-I (they did a culture of the fluid from the sores i had during my initial outbreak). So it is possible to contract HSV-I by asymptomatic asexual contact, so please people talk about this and prevent anyone else from getting it, I'm kind of going through something. Four years ago my love and I both had an OB and when I got the blood work done it came back negative. We were happy in the relationship so I learned to accept it and never really bothered to get tested again. Now, that we broke up I decided to get tested again. I came back negative for both HSV-1 and HSV-2 (got the news!) and have not had an OB since the initial one four years ago. My X on the other hand has had a few small outbreaks and never came back positive for HSV-1.Only the help of Dr Afrid where able to help me out with is Root and Herbs, if you are having any type of this disease or infection like HIV,CANCER OR ANY disease kindly email doctor Afrid for cure:( drafridherbalhome@gmail.com ) or Whatsapp him on +2349057260738.
ReplyDeleteNATURAL HERBS AND VEGANS CURE FOR Herpes 1&2 THAT WORK FAST WITHIN 14 DAYS WITH DR ELINFOH HERBAL MEDICINE, I saw so many testimonies about Dr ELINFOH a great HERBAL DOCTOR that will help you CURE and give you the rightful health to live a joyful life, i didn't believe it at first, but as the pain got worsen and my life was at risk after using lots of ARV drugs from the hospital and no changes so i decided to give a try to Dr Elinfoh I contacted him also and told him i want a cure for Herpes , he gave me advice on what i must do and he deliver it to me in my home address i gave him and i got the medicine which i use according to his instruction, and today i must say I am so grateful to this man Dr Elinfoh for curing me from Herpes and for restoring me back to my normal health and a sound life and i am making this known to every one out there who have been trying all day to be cured from Herpes or any sickness should not waste more time just contact him with his details below, believe me this is the only way to get cured from Hepres , this is the real solution we all have been searching for. Do not waste more time contacting him today for you can also leave a sound and happy life. contact info below. Email: (drelinfohherbalhome@gmail.com ) or WhatsApp or Call him on: +2348110492072. and you can as well contact him on other issues you are having like*HEPATITIS B*DIABETICS*CANCER*ALS*HERPES*BODY REDUCTION*BREAST ENLARGEMENT*FIBROD*CANCER*PREGNANCY MISCARRIAGE*Relationship issues etc.. ** Neoplasms*Cardiovascular Disease (CVD)
ReplyDelete*Chronic Respiratory Diseases
* Mental and Behavioral Disorders
*Alzheimer’s Disease
Email: (drelinfohherbalhome@gmail.com).
Am Laura Mildred by name, i was diagnosed with Herpes 4 years ago i lived in pain with the knowledge that i wasn't going to ever be well again i contacted so many herbal doctors on this issue and wasted a large sum of money but my condition never got better i was determined to get my life back so one day i saw Mr. Morrison Hansen post on how Dr. Emu saved him from Herpes with herbal medicine i contacted Dr. Emu on his Email: Emutemple@gmail.com we spoke on the issue i told him all that i went through and he told me not to worry that everything will be fine again so he prepared the medicine and send it to me and told me how to use it, after 14 days of usage I went to see the doctor for test,then the result was negative, am the happiest woman on earth now thanks to Dr. Emu God bless you. Email him at: Emutemple@gmail.com Call or Whats-app him: +2347012841542
ReplyDelete5 years ago I had warts, I was treated with some liquid applied to the warts they continued to grow and spread... The next 2 doctors did laser surgery to remove them. 1 year after the surgery, they grew back close to where the 1st ones were' so I was finally told it was hpv. I have had it for very long time, I contract it from my cheated boyfriend and I found out he was also infected and I end up the relationship between us. the warts was so embarrasses because it started spreading all over I have be dealing with this things for very long time the last treatment I take was About 2 years ago I applied natural treatment from Dr onokun herbal cure, a week after applying the treatment all the warts was gone. it's now 2 years and some months I don't have single wart or any symptoms of hpv. wow"" it's great, Dr onokun has finally cured me. Anyone living with hpv contact Dr onokun for natural treatment.
ReplyDeleteHis email address: Dronokunherbalcure@gmail.com
I was diagnosed of herpes virus in 2015, I have tried all possible means to get cure but all my effort proved abortive, until a friend of mine introduced me to a herbal doctor called Dr Onokun who prepare herbal medicine to cure all kind of diseases including herpes virus (Hsv), when i contacted this herbal doctor via his email, he sent me herpes virus herbal medicine via courier service, when i received the herbal medicine he gave me step by step instructions on how to apply it, when i applied it as instructed i was totally cured from the virus within 2 weeks of usage. Contact this great herbal doctor today to get your cure.
ReplyDeleteVia Email : Dronokunherbalcure@gmail.com
few weeks ago i left some pains and i want to the hospital my doc told me i had HPV about 1 year and i need to start the treatment early he gave me some medicine to be taking and i told him no i will not because i hated taking medicine when i wont not see cure for the purpose after i got home i started getting worried searching for strong advice and i found valid natural treatment online lot people says they got HPV cure from Dr onokun and i email Dr onokun 3 days passed i purchase his cure online some weeks after the process taking his treatment i got cured i went for checkup twice after taking the natural treatment from the herbalist called Dr onokun and i tested negative contact him on email :Dronokunherbalcure@gmeil.com
ReplyDeleteI actually was told otherwise, that the herpes can not be treated and that there is no way to make it go away once you have it. But I decided to do my own research and found out it is possible to fight it.. I got a lot of information about it here in forums and online,I got Dr Oje Abacha contact on a group were a man from Cordova was sharing his testimony of how he cured him of herpes so I decided to give him a try I emailed him through oje.herbalsolutioncentre@gmail.com . he called me to know if I actually wanted the cured after which the cure was sent to me few days later I was surprised when the same doctor who told me that I was diagnosed of herpes told me that I was completely free from herpes you can reach him through his email oje.herbalsolutioncentre@gmail.com . or through his WhatsApp +2348159734766 if you are diagnosed and also wanna be free from this deadly virus because he is really gonna be of help to you
ReplyDeleteNever give up in life they all say no cure to Herpes Virus which is a big lie I have pass through many process also i never believe there is really cure to HERPES until I meet Dr. Kham the doctor that have been helping many people for many years, I come across this doctor online when I was searching for cure online I found out about this man, and to my greatest surprise this man have the herbal medicine which I have been looking for years I explain my problem to him through the email I found on someone who testify about him also, Dr. Kham write me a reply and explain how the process work so after ordering for the medicine I got it within 4 days and I took it according to the way Dr. Kham instructed, I was so happy after two week I took the medicine there was very big change in my health when I was done with the process I go for test, I found out I am negative that was the day I have the tears of joy you can also get in contact with my doctor through his email now dr.khamcaregiver@gmail.com or you can also WhatsApp him +2348159922297, You can Also Visit his website to know more about him at > https://drkhamherbalhealingcenter.wordpress.com/ or https://drkhamcaregiver.wixsite.com/drkhamcaregiverherba He also have herbs medicine to cured the following diseases; eczema,urethra wart,chronic problems.Herpes, Cancer, Als,Hepatitis, Diabetes, HPV, HIV, Diabetes, Fibroid, Infections,ulcer ETC
ReplyDeleteHappiness is all i see now I never thought that I will be cured from HERPES virus again. I have been suffering from a deadly disease (HERPES) for the past 3 years now, I had spent a lot of money going from one places to another, from churches to churches, hospitals have been my home every day residence. Constant checks up have been my hobby not until this faithful day, I was searching through the internet, I saw a testimony on how DR agaba helped someone in curing his HERPES disease, quickly I copied his email which is Dragabasolutionhome@gmail.com just to give him a test I spoke to him, he asked me to do some certain things which I did, he told me that he is going to provide the herbal cure to me, which he did, then he asked me to go for medical checkup after some days after using the herbal cure, behold I was free from the deadly disease, he only asked me to post the testimony through the whole world, faithfully am doing it now, please brothers and sisters, he is great, I owe him in return. if you are having a similar problem just email him on : Dragabasolutionhome@gmail.com or WhatApp: +2349074536486
ReplyDeleteMy health was horrible before I decided to try the Protocol Of taking Dr Ebhota herbal mixture. I felt there was no hope for my health and I was doubtful to try the Protocol thinking it wouldn’t work because I have visited so many hospital but same result. However, I was convinced by my friend to try the herbal medicine because I wanted to get rid of HPV/WART. The herbal mixture that was given to me was really quick and easy to take, and since I have be taking it for less than 3 days I have less outbreak. But within one week i was fully cured from WART/HPV. The herbal medicine really work and I will like to share this great herb doctor contact with you all email him drebhotasolution@gmail.com or whatsapp +2348089535482. Pls try and help yourself out of warts completely today. he also c My health was horrible before I decided to try the Protocol Of taking Dr Ebhota herbal mixture. I felt there was no hope for my health and I was doubtful to try the Protocol thinking it wouldn’t work because I have visited so many hospital but same result. However, I was convinced by my friend to try the herbal medicine because I wanted to get rid of HPV/WART. The herbal mixture that was given to me was really quick and easy to take, and since I have be taking it for less than 3 days I have less outbreak. But within one week i was fully cured from WART/HPV. The herbal medicine really work and I will like to share this great herb doctor contact with you all email him drebhotasolution@gmail.com o r whatsapp +2348089535482. Pls try and help yourself out of warts completely today. he ure DIABETIES ULCAL CANCER etc.He also told me that he has solution for the flowing.1 Cancer cure2 Diabetes cure3 Ringing ear4 Herpes cure5 Warts cure6 HPV cure7 Get your ex back8 Pregnancy herbal medicine9 Prostate enlargement10 Hepatitis B11 Disability12 Kidney problem Etc.
ReplyDeleteRICK SIMPSON CANNABIS OIL CURES ALL KINDS OF CANCER TOTTALY
ReplyDeleteI am so grateful to Dr. Rick Simpson for providing me with Cannabis oil and Rhino Horns here in the United State of America. I was diagnose with skin cancer 2 years and 3 weeks ago, and ever since then have done a lot of Chemo and Radiation that have not help me, but only damaged my immune system and render me weak and helpless. I came across the Phoenix Tears and i have read about the Hemp oil a lot and saw the Post that Dr. Rick Simpson could provide me with
Cannabis Oil here is the State, I contacted him on: simpsonrick95@gmail.com for the procurement of this medication, to
my surprise the medication was procured and delivered within 48 hours and i have been on treatment for the past 3 months. Am now here to testify that am no longer a cancer patient. I have experience a total transformation in my life with Dr. Rick Simpson Cannabis oil service. Below are the different types of illness he cured
(1) Adenoid Cystic Carcinoma
(2) Anal Cancer (Squamous Cell Carcinoma)
(3) Aplastic Anaemia
(4) Basal Cell Carcinoma
(5) Bladder Cancer
(6) Bone Cancer
(7) Brain Cancer
(8) Breast Cancer
(9) Cervical Cancer
(10) Colon Cancer
(11) Epithelioid Hemangioendothelioma (EHE)
(12) Endometrial Cancer
(13) Kidney Cancer
(14) Leukaemia
(15) Liposarcoma
(16) Liver Cancer
(17) Lung Cancer
(18) Lymphoma
(19) Lymphoma
(20) Melanoma
(21) Neuroblastoma
(22) Ovarian Cancer
(23) Pancreatic Cancer
(24) Parotid Salivary Gland Cancer
(25) Pituitary Gland Tumour
(26) Prostate Cancer
(27) Spinal Tumour
(28) Squamous Cell Carcinoma
(29) Thyroid Cancer
(30) Uterine Cancer
For all cancer patients that lives in American region, Asia, Europe and the world at large, go get your Cannabis oil by contacting: simpsonrick95@gmail.com https://simpsonrick95.over-blog.com/preview/21bb18687f7991d2982c2148291723a576852d1f
I am very grateful to Rick Simpson. My father who is 59 was diagnosed with brain cancer on April 26 2020 and was given 5 months to live. All hope was lost until my friend introduced us to the Rick Simpson cannabis oil treatment. I immediately placed my order for the Rick Simpson oil which was delivered to my address in Viale Roma in 3 days. My Father used the RSO Treatment for 90 days. Now, my Father is completely healed.Email; Ricksimpsoncannabishemoil@gmail.com to get your RickSimpson oil.
ReplyDeletesincerely,libero Stacy
My ex-husband and I had always managed to stay friendly after our divorce in February 2017. But I always wanted to get back together with him, All it took was a visit to this spell casters website last December, because my dream was to start a new year with my husband, and live happily with him.. This spell caster requested a specific love spell for me and my husband, and I accepted it. And this powerful spell caster began to work his magic. And 48 hours after this spell caster worked for me, my husband called me back for us to be together again, and he was remorseful for all his wrong deeds. My spell is working because guess what: My “husband” is back and we are making preparations on how to go to court and withdraw our divorce papers ASAP. This is nothing short of a miracle. Thank you Dr Emu for your powerful spells. Words are not enough. here is his Email: emutemple@gmail.com or call/text him on his WhatsApp +2347012841542
ReplyDeleteHe is also able to cast spell like 1: Lottery 2: Conceive 3: Breakup 4: Divorce 5: Cure for all kinds of diseases and viruses.
I used to suffer from HERPES GENITAL for about 3 years so I can relate to people who have the cure. No medications or treatments ever really did anything for me (i tried them all no one worked. but I was actually able to completely cure HERPES naturally after countless hours of online research. What worked for me is as follows: I saw Dr Oje Abacha Information online that he normally cure and treat disease and infirmities with his Herbal Medicine, I never really believed the people who were recommending him thou i saw on blog about 6 people where also testifying on how they were cured by Dr Oje Abachaand stillEllen recommend Dr Oje Abacha who uses herbal medication to cure HERPES GENITAL VIRUS and gave me his email, so i mail him He told me all the things I need to do and also give me instructions to take, which I Followed properly. Before I knew what is happening after two weeks the Herpes GENITAL VIRUS that were in my body got vanished . so if you are also heart broken and also need a help, you can also email him:oje.herbalsolutioncentre@gmail.com . or through his WhatsApp +2348159734766
ReplyDeleteIf you are infected with any disease like HIV,AIDS,CANCER, LOVE SPELL, HERPES.PVC, or any other disease you can also be happy like me by contacting.....Name: Dr Oje Abacha
herpes is a serious and recurring disease which can't be cured through drugs or injections by the American doctors but the best way to deal with herpes is by taking natural herbs medicine for it and is only few American doctors that know about this herbal medicine from Dr Akhanene .. I have read about Dr Akhanene the great herbalist doctor from African who can cure disease with his powerful herbal medicine. for the people suffering from the following diseases, Herpes, Cancer, Also,Herpatitis, Diabetes, Hps,Infections ETC should contact him for his herbal medicine because i am a living testimony and i was cured of herpes. Although, i sent him what he requested and he sent me his medicine which i took for 1 weeks and today when i went for test i was tested herpes negative. you can reach him through his Emai drakhanenespellhome@gmail.com.com or whatsapp or call him +2348168714427
ReplyDeleteBuy Medicine Online Overnight Delivery USA
ReplyDeleteBuy Xanax Online
Buy Tramadol Online
Buy Ambien Online
Buy Viagra Online
Buy Levitra Online
Buy Cialis Online
Buy Adipex Online
Usapainpharma
Buy Oxycodone Online
Buy Hydrocodone Online
Rite Aid USA offer:
ReplyDelete* No Prescription Required (NO RX)
* Buy Direct and Save Time with Cryptocurrency
* Reshipment is offered if the package does not get to location or refunds
* High-Quality Pharmacy Grade Medicines.
* Best and Affordable prices.
Buy Carisoprodol Online
Buy Soma Online
I think this is a really good topic. You make this information interesting and engaging. You give readers a lot to think about and I appreciate that kind of post.if you are interested to play Satta King game so you can play here Satta King.
ReplyDeleteMedical Pharmacy USA Order Anti-Anxiety, Detoxifying, Pain Relief, Sleeping & Weight Loss Pills online, without prescription at the best price in the United States - Medical Pharmacy USA.
ReplyDeleteBuy Tramadol Online
Buy Hydrocodone Online
Buy Xanax Online
Buy Herbal Product for Granuloma Annulare that encourages you to treat many skin diseases and issues, for example, scabies, rashes, eczema, psoriasis, skin break out and different diseases. Herbs Solutions by Nature uses herbal products and the skin detox formula to cure granuloma annulare naturally. If tiresome these Natural Remedies for Granuloma Annulare you feel unsavory and congenial.
ReplyDeleteAmbien (zolpidem) is among the most popular and effective sedatives that help people fall asleep and keep them asleep for a more extended period. Order Now -
ReplyDeleteBuy Adderall Online
Buy Pain Pills Online
Buy Ambian Online
Buy Ativan Online
Buy Chloroquineonline Online
Buy Codeine Online
Myasthenia Gravis can be treated with the help of natural herbal remedies which are safe to use with no symptom. Buy Herbal Product for Myasthenia Gravis to get rid of this condition. It mostly takes a shot at the underlying driver of disease and manages with the symptoms with the help of herbs. These herbs regulate the tri energies of body which is the leading cause of disease in herbs.
ReplyDeleteOn the happier side though, Natural Treatment for Lipoma has no side effects. In fact, you end up success more health aids and decreasing the risks of getting other health problems. Buy Herbal Product for Lipoma Cure by Exercise Links to an external site. will aid in reducing the risks of circulatory disorders while eating a sufficient amount of vitamins will lessen the risks of getting further common infections like flu.
ReplyDeleteDefinitely and totally cured of herpes virus by Doctor Razor's Quick cure for Herpes. My Sincere Gratitude to Dr Razor, I was infected with HERPES SIMPLEX VIRUS 2 November 13 2018, I went to many hospitals for cure but there was no solution, I was confused because the medical drugs has become my daily food, but i decided to sort after Natural Herbs. One day I sat beside the pool, Browsing and thinking where I could get a solution to end this predicament. I saw a blog on how Dr Razor cured people with his herbal medicine, i did not believe but i just decided to give him a try, Visit Doctor's Razor Website : https://herbalistrazorherb.wixsite.com/drrazorherbalhome . I contacted him and he prepared the herbs for me which I took, and he instructed me to go for a check up, after the test i was confirmed with herpes negative, i am so happy. If you have any problem or you are also infected with any disease, kindly contact him now with his Email: drrazorherbalhome@gmail.com or Whatsapp/call +2349065420442 . .This testimonial serves as an expression of my gratitude.
ReplyDeleteHe also have a herbal cure for COLD SORE,
SHINGLES,
CANCER(Cannabis Oil),
HPV,
ASTHMA,
IMPOTENCE,
BARENESS/INFERTILITY ..
Contact him on this email: drrazorherbalhome@gmail.com , or Whatsapp/call his cell phone number on +2349065420442
Herbal Treatment for Benign Essential Tremor to treat the voice, head, jaw, lips, and face. Herbal Supplement really takes hold and treats the condition naturally. Herbal Treatment for Benign Essential Tremor has been used to treat any kind of tremor, and also nervous restlessness, anxiety, and insomnia for thousands of years, and it continues to be among the most popular Natural Remedies for Benign Essential Tremor to promote relaxation.
ReplyDeleteBuy Herbal Product for Benign Essential Tremor to treat the voice, head, jaw, lips, and face. Herbal Supplement really takes hold and treats the condition naturally.
ReplyDeletehello this is my new site please checkout here by clicking on itkalyanchartresult
ReplyDeleteI was diagnosed as HEPATITIS B carrier in 2013 with fibrosis of the
ReplyDeleteliver already present. I started on antiviral medications which
reduced the viral load initially. After a couple of years the virus
became resistant. I started on HEPATITIS B Herbal treatment from
ULTIMATE LIFE CLINIC (www.ultimatelifeclinic.com) in March, 2020. Their
treatment totally reversed the virus. I did another blood test after
the 6 months long treatment and tested negative to the virus. Amazing
treatment! This treatment is a breakthrough for all HBV carriers.
DO YOU NEED A PERSONAL/BUSINESS/INVESTMENT LOAN? CONTACT US TODAY VIA WhatsApp +19292227023 Email drbenjaminfinance@gmail.com
ReplyDeleteHELLO
Do you find yourself in a bit of trouble with unpaid bills and don’t know which way to go or where to turn? What about finding a reputable Debt Consolidation firm that can assist you in reducing monthly installment so that you will have affordable repayment options as well as room to breathe when it comes to the end of the month and bills need to get paid? Then consider your financial problems over. We Offer LOANS from $3,000.00 Min to $30,000,000.00 Max with a low interest rate of 2% and loan duration of 1 to 35 years to pay back the loan secure and unsecured. Our loans are well insured for maximum security is our priority, Our leading goal is to help you get the services you deserve, Our program is the quickest way to get what you need in a snap. Kindly reduce your payments to ease the strain on your monthly expenses
NO MATTER YOUR CREDIT SCORE. GET YOUR INSTANT LOAN APPROVAL 100% GUARANTEED TODAY VIA WhatsApp:+19292227023 Email: drbenjaminfinance@gmail.com
Nitro Pro Crack With Serial Number 2022 (Torrent)
ReplyDeleteGihosoft iPhone Data Recovery 4.2.8 Crack + Registration Code
DRmare M4V Converter 4.1.1 Crack With Serial Key 2022
Lumion Pro Crack With Activation Code Here 2022
Template Toaster 8.0.0.20695 Crack
Mixcraft 9 Crack With Registration Code Download [Latest]
7-Data Recovery Suite 4.4 Crack + Serial Key Free Download
File Viewer Plus 4.0 Crack With Activation Key
This is very useful software for you and me.
ReplyDeleteI blog often and I genuinely appreciate your content.
xilisoft video converter ultimate crack
tally erp 9 crack
pes 2017 crack
maxbulk mailer pro crack
ReplyDeleteHello, I just discovered your blog on Google and I like it.
it's quite handy. I'll look at the Brussels sprouts.
If you continue to do so in the future, I will appreciate it. Many people will benefit from it.
based on your writing Greetings. Thanks for sharing.
burnintest professional crack
burnintest professional crack
burnintest professional crack
burnintest professional crack
burnintest professional crack
burnintest professional crack
burnintest professional crack
burnintest professional crack
burnintest professional crack
burnintest professional crack
I’ve been surfing on the web more than 3 hours today, yet I never found any stunning article like yours.
ReplyDeleteIt’s alluringly worth for me.
As I would see it, if all web proprietors and bloggers made puzzling substance as you did.
the net will be in a general sense more beneficial than at whatever point in late memory.
hdrsoft photomatix pro crack
faststone image viewer crack
fonepaw iphone data recovery crack
auslogics bitreplica crack
rollback rx crack
hasleo data recovery crack
Hi! Your Blog Writing is Very Wonderfull And Amazing. Thanks For Sharing...
ReplyDeleteformat factory crack
freemake video converter crack
iobit uninstaller crack serial key
bandicam crack
halftime vst crack latest version
1. Blueberry Tea in Pakistan|03016690333
ReplyDelete2. Green World Herbs Toothpaste in Pakistan|03016690333
3. Green World Detoxin Pad in Pakistan|03016690333
4. Green World Olive Soap in Pakistan|03016690333
5. Cell Phone Radiation Protection Paster in Pakistan|03016690333
6. Asthijivak in Pakistan|03016690333
7. Breast Enlargement Cream in Pakistan|03016690333
8. Dempo Snow Cream in Pakistan|03016690333
9. Dexe Black Hair Shampoo in Pakistan|03016690333
10. Eco Slim in Pakistan|03016690333
11. Vital Honey in Pakistan|03016690333
WhatsApp Call & Sms 03016690333
Home Delivery Available in Pakistan
Availability: In stock
Basic benefits of massage therapy is the increased circulation of the blood in any area that is massaged and that increase in blood flow naturally aids in the healing process. 강북구출장안마
ReplyDeleteYou are so interesting! I don't think I've read anything like this before.
ReplyDeleteIt's great to find someone with real ideas on this topic. Indeed ... thank you very much for starting.
This site is something needed on the internet, not real!
This is a great blog! Your site is loading too fast!
What type of web server do you use? Can you send me an affiliate link for your web host?
plum amazing iwatermark pro crack
program4pc audio converter pro crack
aimersoft dvd creator crack
wondershare dvd creator crack
winext batch operator enterprise crack
treesize professional crack
softmaker flexipdf 2019 professional crack
viewcompanion premium crack
red gate smartassembly crack
Agen Daftar Sbobet88 Pakai Gopay
ReplyDeleteDownload Apk Slot Joker
Daftar IDN Poker Tanpa Rekening
Apk IDN Poker
Login Club388
Deposit IBCBET
Nice & Impressive Blog!https://trustedcrack.com/pgware-pcboost-crack/
ReplyDeleteI am living testimony..
ReplyDeleteA friend of mine recommended me to contact this herbal Doctor for herpes cure and he asked me to purchase his herbal medicine which i did, when i received this herbal medicine, he gave me instructions on how to use it, after taking the medicine as instructed for 2 weeks, i went for check up and the result shows negative and i was cured of herpes, I am now free from Herpes. You can contact him on his email …………His result is 💯💯💯💯💯 guaranteed. I highly recommend..........
You can win your Ex lover back in 48 hours just like me.
Very effective ...
For fast and reliable solution.
Get boyfriend back after break up.
Get girlfriend back after break up.
Get Gay partner back.
Get Lesbian partner back.
Make Your Husband/Wife love you Forever.
Stop Having Bad Dreams.
Make Women/Men To Run After You.
Stop Divorce from happening.
Divorce Your Husband/Wife.
Make Partner marry you.
Make Wishes To Be Granted.
Win Court Case/Law suit.
Get pregnant.
Luck To Win A Lottery.
Stop Marriage/Relationship from Breaking Apart.
I used his herbal remedy to cure Hsv-2..
100% result guaranteed..
Email Robinsonbucler@gmail com………
PC Privacy Shield Crack is outstanding software seems to have electronic substantial draw, include precedent surfing action also check information, will be removed through over creation.
ReplyDeletePC Privacy Shield Crack
Kinemaster pro crack is a open, untie-basis substitute to Full edition expert. It is certainly a dissimilar KineMaster version. “Efficient” and “break” Android is what Pub movable place for. A fresh alternative of that an original machine is referred to as a mudded.
ReplyDeleteKinemaster Pro Crack
WhatsApp for Windows Crack serving the consumer might attach also does what consumer desire, wherever exterior the home, whether several mobile, and desktop, gratitude to scrap official 256-bit application.
ReplyDeleteWhatsApp For Windows Crack