Alpha-thalassemia (α-thalassemia, α-thalassaemia) is a form of thalassemia involving the genes HBA1 and HBA2. Alpha-thalassemia is due to impaired production of 1,2,3, or 4 alpha globin chains, leading to a relative excess of beta globin chains. The degree of impairment is based on which clinical phenotype is present (how many chains are affected).
Epidemiology

The worldwide distribution of inherited alpha-thalassemia corresponds to areas of malaria exposure, suggesting a protective role for alpha-thalassemia against the more severe manifestations of malaria. Thus, alpha-thalassemia is common in sub-Saharan Africa, the Mediterranean Basin, the Middle East, South Asia, and Southeast Asia, and different genetic subtypes have variable frequencies in each of these areas. The epidemiology of alpha-thalassemia in the US reflects this global distribution pattern. The most common form of alpha(+) thalassemia seen in the US is due to the -alpha(3.7) deletion, a single alpha-globin gene deletion, and is present in approximately 30% of African Americans. However, even in the homozygous state this disorder will result only in a mild microcytic anemia. The more serious clinical disorders of Hb H and Hb Bart hydrops fetalis syndrome, although found throughout the US today, are more common in the Western US and have dramatically increased in prevalence in the past 2 decades due to increased Asian immigration.
Causes

It is most commonly inherited in a Mendelian recessive fashion. It is also connected to the deletion of the 16p chromosome.
It can also be acquired, under rare circumstances. Due to the low occurrence of alpha-thalassemia, the disease can be mistaken for iron deficiency anemia.
Pathophysiology
α thalassemias result in decreased alpha-globin production, therefore fewer alpha-globin chains are produced, resulting in an excess of β chains in adults and excess γ chains in newborns. The excess β chains form unstable tetramers (called Hemoglobin H or HbH of 4 beta chains) which have abnormal oxygen dissociation curves. The excess γ chains form tetramers which are poor carriers of O2 since their affinity for O2 is too high so it is not dissociated in the periphery. Homozygote α0 thalassaemias, where there is lots of γ4 but no α-globins at all (referred to as Hb Barts), often result in still birth.
Types

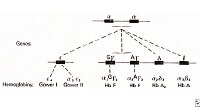
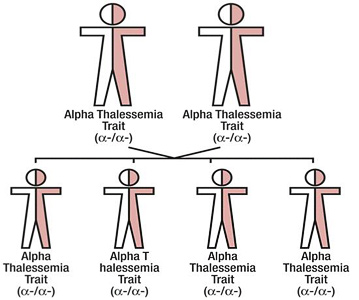
There are two genetic loci for α globin, and thus four genes in diploid cells. Two genes are maternal in origin and two genes are paternal in origin. The severity of the α thalassemias is correlated with the number of affected α globin genes: the greater, the more severe will be the manifestations of the disease.
When noting the genotype, a "-" indicates an absence of function, and a "α" indications a functional alpha chain. (In contrast to the "βo" and "β+" notation used in beta-thalassemia, in alpha-thalassemia a distinction between absent and reduced function is not usually noted.)
References





Go for the original. Piracy is an alarming illegal practice in the Philippines. As consumers, the best thing that we could do is to give our sincerest support through buying original CDs and music video recordings of local artists. Nowadays, the web has put more pressure on the music industry since people can easily access songs by means of Pinoy Tv Online Video downloading and file sharing.
ReplyDeleteI was suffering from Warts Simplex Virus, i was totally depressed due to my predicament , until i meet Dr Nana the great Traditional healer who cured me. you can also contact him now on whatsapp +2347014784614.or via email:on drnanaherbalsolutionhome@gmail .com for more information.
DeleteI was suffering from Warts Simplex Virus, i was totally depressed due to my predicament , until i meet Dr Nana the great Traditional healer who cured me. you can also contact him now on whatsapp +2347014784614.or via email:on drnanaherbalsolutionhome@gmail .com for more information.
I was suffering from Warts Simplex Virus, i was totally depressed due to my predicament , until i meet Dr Nana the great Traditional healer who cured me. you can also contact him now on whatsapp +2347014784614.or via email:on drnanaherbalsolutionhome@gmail .com for more information.
Hello everyone, am very happy to share this little awesome testimony about Dr olu a great herbal doctor who help me enlarge my penis size.3.2 cm to 8.3 cm longer with his herbal cream mixture, my girlfriend is now so amazed with the autonomous size of my penis , if you you are also in need of help on how to enlarge your penis to become bigger and stronger I adverse you to contact Dr on his email (drolusolutionhome@gmail.com) ) you or contact on whatsapp number +2348140654426 because he is one of the best herbal doctor that i can only show you up to, if your penis is 4.2 cm and want to get it reach 9.2 cm within three weeks i Dr olu is also specialized on obey m breast and boobs enlargement i advise you to contact him for help
Deletei already gave up on ever getting cured of HSV2 because i have try many treatment none of them work out for me i have go to different hospital they always tell me same thing there is no cure for herpes when i came across a post about Dr ohunyom, in the net from a lady called Angela i contacted him and he reassured me with him herbal medicine which i took according to the way he instructed, that how i was cured. I doubted at first because i have been to a whole lot of reputable doctors, tried a lot of medicines but none was able to cure me. so i decided to listen to him and he commenced treatment, and under two weeks i was totally fee from #Herpes. i want to say a very big thank you to DR ohunyom for what he has done in my life. feel free to leave him a message on email drohunyom@gmail.com and also WhatsApp him +2349060579973..
DeleteHe can still be able to help you with this herbs medicine:
1...ALS CURE/DIABETES CURE/EPILESY/HPV CURE/LUPUS/HEPATITIS/CANCER/GOUT
I have used Dr Aziba herbal Medicine cure remedy for 2 WEEKS now. So far, it has cured my HERPES, eliminated my COLD SORE, improved energy and metabolism, in the morning I feel recharged and I no longer feel sleepy after a meal. As for the scent, it does smell like motor oil but I chase it down with 100% Herbal medicine from Dr Aziba. twice a day AFTER a meal. I also experienced vivid dreams for about 5 days now it’s diminishing to during the first 2 or 3 hours of sleep . contact Dr Aziba: EMAIL: priestazibasolutioncenter@gmail.com or whatsapp +2348100368288 and He also have herbs medicine to cure some popular diseases....
DeleteSome other areas of your health you may need Dr Aziba help to get cured.
{1}Diabetes
{2}HPV
{3}ALS
{4}Herpes
Contact
Dr via Email: Priestazibasolutioncenter@gmail.com
WhatsApp : +2348100368288
God bless Dr ojoka for his marvelous work in my life, I was diagnosed of HERPES since 2011 and I was taking my medications, I wasn't satisfied i needed to get the HERPES out of my system, I searched about some possible cure for HERPES i saw a comment about Dr ojoka how he cured HERPES with his herbal medicine, I contacted him and he guided me. I asked for solutions, he started the remedy for my health, he sent me the herbal medicine through UPS SPEED POST. I took the medicine as prescribed by him in 2 weeks and 3 days later i was cured from HERPES, Dr ojoka truly you are great, do you need his help also? Why don't you contact him through email:{dr.ojokarootandherbal@gmail.com or whats app/call +2348144172934} check his blog: https://perfectherbalcure.blogspot.com/ check his blog: https://penisherbalenlarge.blogspot.com/ FB page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/
DeleteGod bless Dr ojoka for his marvelous work in my life, I was diagnosed of HERPES since 2011 and I was taking my medications, I wasn't satisfied i needed to get the HERPES out of my system, I searched about some possible cure for HERPES i saw a comment about Dr ojoka how he cured HERPES with his herbal medicine, I contacted him and he guided me. I asked for solutions, he started the remedy for my health, he sent me the herbal medicine through UPS SPEED POST. I took the medicine as prescribed by him in 2 weeks and 3 days later i was cured from HERPES, Dr ojoka truly you are great, do you need his help also? Why don't you contact him through email:{dr.ojokarootandherbal@gmail.com or whats app/call +2348144172934} check his blog: https://perfectherbalcure.blogspot.com/ check his blog: https://penisherbalenlarge.blogspot.com/ FB page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/
God bless Dr ojoka for his marvelous work in my life, I was diagnosed of HERPES since 2011 and I was taking my medications, I wasn't satisfied i needed to get the HERPES out of my system, I searched about some possible cure for HERPES i saw a comment about Dr ojoka how he cured HERPES with his herbal medicine, I contacted him and he guided me. I asked for solutions, he started the remedy for my health, he sent me the herbal medicine through UPS SPEED POST. I took the medicine as prescribed by him in 2 weeks and 3 days later i was cured from HERPES, Dr ojoka truly you are great, do you need his help also? Why don't you contact him through email:{dr.ojokarootandherbal@gmail.com or whats app/call +2348144172934} check his blog: https://perfectherbalcure.blogspot.com/ check his blog: https://penisherbalenlarge.blogspot.com/ FB page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/
Traditional Herbal doctor Sayo Cure HSV-2 Disease
DeleteI want to thank Dr Sayo for helping me with his Herbal Medicine that cure my HSV-2 Disease completely.i have been living with HSV-2 Disease for the past 3yrs. i was so happy when i found DrSayo blogs: how Dr Sayo help people in the world.to cure HSV, with Herbal Medicine he Cured My HSV -2 Negative after using the Herbal Medicine Dr Sayo sent to me. I give thanks to God and healed completely. you are the best Traditional Herbal doctor in the world Dr Sayo, Contact Doctor Sayo Email sayoherbalhealer@gmail. com OR Contact Sayo by check FB Page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/ check website; http://sayoherbalhealer.website2.me/ check blog: https://sayoherbalhealer.blogspot.com/
Traditional Herbal doctor Sayo Cure HSV-2 Disease
I want to thank Dr Sayo for helping me with his Herbal Medicine that cure my HSV-2 Disease completely.i have been living with HSV-2 Disease for the past 3yrs. i was so happy when i found DrSayo blogs: how Dr Sayo help people in the world.to cure HSV, with Herbal Medicine he Cured My HSV -2 Negative after using the Herbal Medicine Dr Sayo sent to me. I give thanks to God and healed completely. you are the best Traditional Herbal doctor in the world Dr Sayo, Contact Doctor Sayo Email sayoherbalhealer@gmail. com OR Contact Sayo by check FB Page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/ check website; http://sayoherbalhealer.website2.me/ check blog: https://sayoherbalhealer.blogspot.com/
Traditional Herbal doctor Sayo Cure HSV-2 Disease
I want to thank Dr Sayo for helping me with his Herbal Medicine that cure my HSV-2 Disease completely.i have been living with HSV-2 Disease for the past 3yrs. i was so happy when i found DrSayo blogs: how Dr Sayo help people in the world.to cure HSV, with Herbal Medicine he Cured My HSV -2 Negative after using the Herbal Medicine Dr Sayo sent to me. I give thanks to God and healed completely. you are the best Traditional Herbal doctor in the world Dr Sayo, Contact Doctor Sayo Email sayoherbalhealer@gmail. com OR Contact Sayo by check FB Page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/ check website; http://sayoherbalhealer.website2.me/ check blog: https://sayoherbalhealer.blogspot.com/
I Never believed i was ever going to be HSV FREE again, DR.Ehiaguna has given me reasons to be happy, i was HSV positive for 2 years and all the means and medicine i tried for treatment was not helpful to me, but when i came on the Internet i saw great testimony about DR.Ehiaguna on how he was able to cure someone from HSV 2, this person said great things about this man, and advice we contact him for any Disease problem that DR.Ehiaguna can be of help, well i decided to give him a try, he requested for my information which i sent to him, and he told me he was going to prepare for me a healing portion, which he wanted me to take for days, and after which i should go back to the hospital for check up, well after taking all the treatment sent to me by DR.Ehiahuna , i went back to the Hospital for check up, and now i have been confirmed HSV NEGATIVE, friends you can reach DR.Ehiaguna on any treatment of any Disease cos i saw many testimony of different disease like, HEPATITIS,HIV AIDS,EPILEPSY, CANCER,CFS he is the one only one i can show you all up to, reach him on drehiaguna@gmail.com or whatsApp him now +2348073908953. quick contact him for help and you can just quickly drop your number on your first mail as i did for easily conversation
DeleteNever give up in life they all say no cure to HSV 2 which is a big lie I have pass through many process also i never believe there is really cure to HERPES until I meet Dr. Sayo the doctor that have been helping many people for many years, I come across this doctor online when I was searching for cure online I found out about this man, and to my greatest surprise this man have the herbal medicine which I have been looking for years I explain my problem to him through the email I found on someone who testify about him also, Dr.Sayo reply and explain how the process work so after ordering for the medicine I got it within 4 days and I took it according to the way Dr.Sayo instructed, I was so happy after two week I took the medicine there was very big change in my health when I was done with the process I go for test, I found out I am negative that was the day I have the tears of joy you can also get in contact with my doctor through his email now (sayoherbalhealer@gmail. com) OR Contact Sayo by check FB Page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/ check website; http://sayoherbalhealer.website2.me/ check blog: https://sayoherbalhealer.blogspot.com/ And He also have herbs medicine to cured the following diseases; eczema,urethra wart,chronic problems.Herpes, Cancer, Als,Hepatitis, Diabetes, HPV,Infections,ulcer ETC
DeleteNever give up in life they all say no cure to HSV 2 which is a big lie I have pass through many process also i never believe there is really cure to HERPES until I meet Dr. Sayo the doctor that have been helping many people for many years, I come across this doctor online when I was searching for cure online I found out about this man, and to my greatest surprise this man have the herbal medicine which I have been looking for years I explain my problem to him through the email I found on someone who testify about him also, Dr.Sayo reply and explain how the process work so after ordering for the medicine I got it within 4 days and I took it according to the way Dr.Sayo instructed, I was so happy after two week I took the medicine there was very big change in my health when I was done with the process I go for test, I found out I am negative that was the day I have the tears of joy you can also get in contact with my doctor through his email now (sayoherbalhealer@gmail. com) OR Contact Sayo by check FB Page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/ check website; http://sayoherbalhealer.website2.me/ check blog: https://sayoherbalhealer.blogspot.com/ And He also have herbs medicine to cured the following diseases; eczema,urethra wart,chronic problems.Herpes, Cancer, Als,Hepatitis, Diabetes, HPV,Infections,ulcer ETC
The doctors said Herpes virus do not have medical cure because the virus is capable of hiding within the human cells, it remains protected from your immune system. Herpes isn’t a special virus – your immune system has the tools to fight it back. But because it is able to lay dormant in protected cells, your immune system is unable to remove it from your body,But with strong reactive herbal medication is capable of getting rid of the virus gradually and totally from your body without damaging any of your cells,natural herbs kills the virus totally not just reducing the out break. Get natural herbs cure Email DR. VOODOO at voodoospelltemple66@gmail.com Add Dr voodoo on whatsApp +2348140120719
DeleteHERPES CURE....THIS IS REAL ! TAKE TIME TO READ
DeleteTESTIMONY OF HOW I GOT CURED FROM GENITAL HERPES SIMPLEX VIRUS WITHIN 14DAYS[with Herbal Traditional medicine]
Hello Everyone out there, I'm Toshia Cook, from Texas USA. I am here to give my testimony about a Herbalist called Dr SAYO, who helped me in my life. I was infected with HERPES SIMPLEX VIRUS 2 in 2010, i went to many hospitals for cure but there was no solution, so I was thinking how can I get a solution out so that my body can be okay. One day I was at the river side thinking where I could get solution. so a lady walked to me asking me why I was so sad and i opened up all to her telling her my problem, she told me that she could help me out, she introduced me to a doctor who uses herbal medication to cure HERPES SIMPLEX VIRUS 2 and gave me his email, so i mailed him. He told me all the things I needed to do and also gave me instructions, which I followed properly. Before I knew what is happening after two weeks the HERPES SIMPLEX VIRUS that was in my body got vanished . so if you are also heart broken and also need a help, you can also email him via: (sayoherbalhealer@gmail .com)
He also have a herbal cure for HIV,
CANCER,
ASTHMA,
IMPOTENCE,
BARENESS/INFERTILITY ...
CURE YOUR SELF FROM THAT ILLNESS TODAY,DON'T SPREAD YOUR INFECTION OR DISEASES TO OTHER PEOPLE.
Contact him today and you will have a testimony...Good luck
HERPES CURE....THIS IS REAL ! TAKE TIME TO READ
DeleteTESTIMONY OF HOW I GOT CURED FROM GENITAL HERPES SIMPLEX VIRUS WITHIN 14DAYS[with Herbal Traditional medicine]
Hello Everyone out there, I'm Toshia Cook, from Texas USA. I am here to give my testimony about a Herbalist called Dr SAYO, who helped me in my life. I was infected with HERPES SIMPLEX VIRUS 2 in 2010, i went to many hospitals for cure but there was no solution, so I was thinking how can I get a solution out so that my body can be okay. One day I was at the river side thinking where I could get solution. so a lady walked to me asking me why I was so sad and i opened up all to her telling her my problem, she told me that she could help me out, she introduced me to a doctor who uses herbal medication to cure HERPES SIMPLEX VIRUS 2 and gave me his email, so i mailed him. He told me all the things I needed to do and also gave me instructions, which I followed properly. Before I knew what is happening after two weeks the HERPES SIMPLEX VIRUS that was in my body got vanished . so if you are also heart broken and also need a help, you can also email him via: (sayoherbalhealer@gmail .com) OR Contact Sayo by check FB Page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/ check blog: https://sayoherbalhealer.blogspot.com/
He also have a herbal cure for HIV,
CANCER,
ASTHMA,
IMPOTENCE,
BARENESS/INFERTILITY ...
CURE YOUR SELF FROM THAT ILLNESS TODAY,DON'T SPREAD YOUR INFECTION OR DISEASES TO OTHER PEOPLE.
Contact him today and you will have a testimony...Good luck
The doctors said Herpes virus do not have medical cure because the virus is capable of hiding within the human cells, it remains protected from your immune system. Herpes isn’t a special virus – your immune system has the tools to fight it back. But because it is able to lay dormant in protected cells, your immune system is unable to remove it from your body,But with strong reactive herbal medication is capable of getting rid of the virus gradually and totally from your body without damaging any of your cells,natural herbs kills the virus totally not just reducing the out break. Get natural herbs cure Email DR. VOODOO at voodoospelltemple66@gmail.com Add Dr voodoo on whatsApp +2348140120719
Delete"He has learned a big lesson out of that. He became more cautious and has matured a lot as a fighter," says Restituto "Buboy" Fernandez, a Watch Pinoy Tv Shows Live long-time associate and the assistant trainer for Pacquiao.
ReplyDeleteBelieve me I have used Dr Aziba herbal Medicine cure remedy for 2 WEEKS now. So far, it has cured my HERPES, eliminated my COLD SORE, improved energy and metabolism, in the morning I feel recharged and I no longer feel sleepy after a meal. As for the scent, it does smell like motor oil but I chase it down with 100% Herbal medicine from Dr Aziba. twice a day AFTER a meal. I also experienced vivid dreams for about 5 days now it’s diminishing to during the first 2 or 3 hours of sleep . contact Dr Aziba: EMAIL: priestazibasolutioncenter@gmail.com or whatsapp +2348100368288 and He also have herbs medicine to cure some popular diseases....
DeleteSome other disease disturbing your health get connected with Dr Aziba for the herbal cure
{1}Diabetes
{2}HPV
{3}ALS
{4}Penis Enlargement
{5}ECZEMA
And Lots More...
Contact
Dr via Email: Priestazibasolutioncenter@gmail.com
WhatsApp : +2348100368288
I'm so exited to share my testimonies about the Good Work of Dr. Udebhu who get me cured from herpes simplex virus (HSV1&2) with his herbs, I never thought that I will live on earth before the year runs out. I have been suffering from herpes, I had spent a lot of money going from one Hospital to another looking for way to get rid of this disease, the hospital have been my home everyday residence. Constant checks up have been my hobby not until this faithful day, I was searching
Deletethrough the internet, I saw a testimony on how Dr.Udebhu helped someone in curing his herpes disease using his healing
Herbs, quickly I copied his email just to give him a test I spoke to him, he told me that he is going to provide the herbal cure to me, which he
did, after i receive his herbs and i take it as instructed i was cure permanently from herpes my herpes disease was gone. so I decided to share my testimony, that nothing is impossible with God, God use a man to healed me. No matter what you are passing through, no matter how deadly the sickness is and no matter what the situation is God that did mine is still going to do yours, people suffering from herpes, brain tumor, kidney disease, pcos, AIDS, ALS,copd, asthma, athritis,herpes, Cancer,Hpv, any kind of disease, you can rich him now via ? Gmail address: drudebhuherbalhome@gmail.com or whatsapp +2349051254152
Duterte has repeatedly warned he may impose martial law, saying military rule could solve many of the Pinoy Tv live Talk Show.
ReplyDeleteThanks to Internet technology, the world of advertising has gone to a whole new level. No longer would you need to buy traditional magazines to search for things to buy, look for jobs or to advertise. You can simply check out Online classified ad websites and you can find the information that you need in no time at all. And most of all, most of Pinoy Channel Online Videos classified ad websites do not charge for usage.
ReplyDeleteEven though mobile analog Pinoy Tv Latest Video Online receivers has been in the marketplace for several years they have not been a big triumph, which may be due to the fact that they do not have any new features over the normal Television. In a far-ahead scenario, the mobile terminal could merge with the remote controller of the television, taking over the interactive parts of television viewing.
ReplyDeleteOther popular children's programs with educational value alongside the entertainment factor include Thomas the Tank Engine & Friends, Barney and Friends, which deals mainly with personal relationships and self-esteem, and the long-running Blue's Clues, which has a heavy emphasis on counting, colors, shapes, and the alphabet for very young watchers. The Magic School Bus is television with a science twist for a slightly older audience, following the adventures of a science class and their crazy escapades under the direction of Pinoy Tv HD Tube their wacky teacher.
ReplyDeleteI can’t believe this. A great testimony that i must share to all HERPES SIMPLEX VIRUS patient in the world i never believed that their could be any complete cure for Herpes or any cure for herpes,i saw people’s testimony on blog sites of how dr Ero prepare herbal medicine that cure and brought them back to life again. i had to try it too and you can,t believe that in just few weeks i started using it all my pains stop gradually and i had to leave without the pills the doctor gave to me. Right now i can tell you that few months now i have not had any pain,delay in treatment leads to death. Here is his email:(dreroherbaltreatment@gmail.com) whatsapp him with +2348073673757 or text/call me 650) 653-8578....
ReplyDeleteI was diagnosed of Herpes in 2014 and I have tried all possible means to get the cure but all to no avail, until i saw a post in a health forum about a Herbal Doctor(Dr Aloha) who prepares herbal medicine to cure all kind of diseases including Herpes, at first i doubted if it was real but decided to give it a try, when i contacted Dr Aloha through his Email: Alohaherbalhome@gmail.com he guided me and prepared a herbal medicine and sent it to me via courier Delivery service,when i received the package (herbal medicine) He gave me instructions on how to consume it,i started using it as instructed and i stooped getting outbreaks and the sores started vanishing, could you believe i was cured of this deadly virus within 10-14 days of using this REMEDY,couldn't believe the healing at first until i went for a check-up,indeed i was tested negative to HERPES. Contact this great herbal Doctor today via Email: Alohaherbalhome@gmail.com and get cured permanently. or Call/Text: +1(312) 361-5917
ReplyDeleteDr. Osagie God will continue to bless you more abundantly, for the good works you are doing in peoples life, I will keep on writng and posting testimonies about you on the Internet, I’m John Homles , I was a HIV patient, I saw a blog on how Dr. Osagie cured someone, I contacted him and also got my healing, kindly email him now on (drosagiesolutiontemple@gmail.com OR
ReplyDeleteDROSAGIESOULTIONTEMPLE@YAHOO.COM ) or His Whatsapp number: +2347030465649...thank you my great DR
Dr OSAGIE CAN AS WELL CURE THE FOLLOWING DISEASE:-
HIV/AIDS
HERPES
CANCER
i am just using this time to thanks Stella Rams for posting the number of the good man Mr Ero and am thanking Mr Ero also, i don't have any bad pap smear again ,the drugs work like magic ,if anybody want good cure of HPV , i will advice you to text or call me on 270) 693-5854 for more information or you can contact dr on dreroherbaltreatment@gmail.com his herbal is the best as far as I'm concerned ,thanks once again.
ReplyDeleteAs you all know that HIV/AIDs and Cancer are getting stronger and spreading day by day and that we are not responsible for the pains of our loved ones, but it’s our responsibility to help those who are infected with viruses. The only way for us to find peace in this world is to love one another and stand by each other. Am sharing this comment because I was once suffering from HIV for 4 years and Genital Herpes for some months until I got help from Dr. Osojo who helped me with a herbal cure for both Genital Herpes and my 4 years HIV virus. If you also need help, you can also reach him: { dr.osojoherbalcureofalldisease@gmail.com } or call him on +234 810 066 3964. You could also use this link, http://drosojoherbalcure.wixsite.com/ofalldisease .Having HIV/Aids or Herpes isn’t the end of your life, it’s actually not a death sentence for you, there is a cure and Dr. Osojo can help you with it as he did mine.
ReplyDeleteHello everyone, I was infected with the herpes virus and I got cured from the herpes virus few months ago after i contacted Dr Azuka. I saw a post on the internet after i have seek healing for several years from different doctors in California. I sent the Doctor a request for help, just a few email i followed his instruction and he sent me the medication. Now i am negative and i referred him to all my friend who had this same sickness and they have gotten their cure too. You can contact him via his email: Dr.azukasolutionhome@gmail.com or Phone call or whatsAPP: +2348132777335. While i was on his medication i understood that he can also cure HERPES, HIV/AIDS, CANCER, Male/female menopause, Miscarriage, Menstruation problems, PREGNANCY PROBLEM, EPILEPSY, GONORRHEA, LASSA FEVER, OBESITY, KIDNEY FAILURE, HYPERTENSION, FIBROID TUMOR, BRAIN FOG, EPSTEIN-BARR VIRUS and many more.
ReplyDeleteContact him today and get your problem solved.
Hello everyone, I was infected with the herpes virus and I got cured from the herpes virus few months ago after i contacted Dr Azuka. I saw a post on the internet after i have seek healing for several years from different doctors in California. I sent the Doctor a request for help, just a few email i followed his instruction and he sent me the medication. Now i am negative and i referred him to all my friend who had this same sickness and they have gotten their cure too. You can contact him via his email: Dr.azukasolutionhome@gmail.com or Phone call or whatsAPP: +2348132777335. While i was on his medication i understood that he can also cure HERPES, HIV/AIDS, CANCER, Male/female menopause, Miscarriage, Menstruation problems, PREGNANCY PROBLEM, EPILEPSY, GONORRHEA, LASSA FEVER, OBESITY, KIDNEY FAILURE, HYPERTENSION, FIBROID TUMOR, BRAIN FOG, EPSTEIN-BARR VIRUS and many more.
ReplyDeleteContact him today and get your problem solved.
Happiness is all i see now I never thought that I will be cured from HERPES virus again. I have been suffering from a deadly disease (HERPES) for the past 3 years now, I had spent a lot of money going from one places to another, from churches to churches, hospitals have been my home every day residence. Constant checks up have been my hobby not until this faithful day, I was searching through the internet, I saw a testimony on how Dr Ogudugu helped someone in curing his HERPES disease, quickly I copied his email which is greatogudugu@gmail.com just to give him a test I spoke to him, he asked me to do some certain things which I did, he told me that he is going to provide the herbal cure to me, which he did, then he asked me to go for medical checkup after some days after using the herbal cure, behold I was free from the deadly disease, he only asked me to post the testimony through the whole world, faithfully am doing it now, please brothers and sisters, he is great, I owe him in return. If you are having a similar problem just email him on ( greatogudugu@gmail.com ) or you can whatsApp his mobile number on +27663492930
ReplyDeleteHello Everybody,
ReplyDeleteMy name is Mrs Sharon Sim. I live in Singapore and i am a happy woman today? and i told my self that any lender that rescue my family from our poor situation, i will refer any person that is looking for loan to him, he gave me happiness to me and my family, i was in need of a loan of $250,000.00 to start my life all over as i am a single mother with 3 kids I met this honest and GOD fearing man loan lender that help me with a loan of $250,000.00 SG. Dollar, he is a GOD fearing man, if you are in need of loan and you will pay back the loan please contact him tell him that is Mrs Sharon, that refer you to him. contact Dr Purva Pius,via email:(urgentloan22@gmail.com) Thank you.
I made a promise to tell my testimony all over the globe once my man come back to me and my husband came just as Great Mother told me when she helped me. I made the right choice to have contacted Great Mother the great spell caster who is capable of helping you solve your problems. My man left me to suffer and i never believed that he will come back to me again but when i contacted DR ISIKOLO she assured me that my man will come again. She gave me some list of items to buy but i could not get them in my country and so i sent her the money and she bought the items and prepared the perfect love spell. My husband ran back to me in 3 days time begging for forgiveness THIS IS SO REAL AND GENUINE. NO SCAMS. I was so surprise that the spell worked just as she told me. This is so amazing and i want you to believe because Great Mother is so real and i urge you contact her now on his email so that he can also help you the way she helped me. His email is isikolosolutionhome@gmail.com. call or Whatsapp him on +2348133261196
ReplyDelete
ReplyDeleteHi Friends....I just want to say a big thanks to ( DR AZUKA ) for what he has done for me and my family by helping me to win lottery. I won Five Hundred and Thirty Three Million Us Dollars. (($533,000,000.00)). His lottery spell is the best and so amazing. My life is now balance and i am Now Very Rich As i have always wanted and i am out of doubt. Are you looking for a real and genuine spell caster to help you win big in any kind of lottery you play? you can contact Dr.Azuka , today all your plan to ever win Big in Lottery will Come True and work out well for you. His spell very real and genuine i can not still believe it And His spell also work very fast. Thank you so much Dr. Azuka for dedicating your time to cast the Lottery spell for me. I got my bills paid and my debt cleared and am still mega rich. If you need a real And Genuine spell aster to help you Win lottery,kindly contact him for fast and urgent Help via email : Dr.azukasolutionhome@gmail.com or Whatsapp or Call Via +2348132777335
i have been suffering from (HERPES) disease for the last four years and had constant pain, especially in my knees. During the first year,I had faith in God that i would be healed someday.This disease started circulate all over my body and i have been taking treatment from my doctor, few weeks ago i came on search on the internet if i could get any information concerning the prevention of this disease, on my search i saw a testimony of someone who has been healed from (Hepatitis B and Cancer) by this Man Dr Isibor and she also gave the email address of this man and advise we should contact him for any sickness that he would be of help, so i wrote to Dr Isibor telling him about my (HERPES Virus) he told me not to worry that i was going to be cured!! hmm i never believed it,, well after all the procedures and remedy given to me by this man few weeks later i started experiencing changes all over me as the Dr assured me that i have cured,after some time i went to my doctor to confirmed if i have be finally healed behold it was TRUE, So friends my advise is if you have such sickness or any other at all you can email Dr Isibor on his email :drisiborspellhome@gmail.com or his whatsApp contact +2348107855231.
ReplyDeleteI am so happy, i never believe i will be this happy again in life, I was working as an air-hoster ( cabby crew ) for 3years but early this year, i loose my job because of this deadly disease called Herpes virus (HSV), I never felt sick or have any symptom, till all workers were ask to bring their doctors report, that was how i got tested and i found out that am HSV positive that make me loose my job, because it was consider as an STD and is incurable disease, i was so depress was thinking of committing suicide, till i explain to a friend of mine, who always said to me a problem share is a problem solved, that was how she directed me to Dr Isibor, that was how i contacted him and get the medication from this doctor and i got cured for real, I just went back to my work and they also carry out the test to be real sure and i was negative. Please contact this doctor if you are herpes positive diseases his email is: drisiborspellhome@gmail.com or you can call or whatsApp his mobile number on +2348107855231.
ReplyDeleteI was suffering from Warts Simplex Virus, i was totally depressed due to my predicament , until i meet Dr Nana the great Traditional healer who cured me. you can also contact him now on whatsapp +2347014784614.or via email:on drnanaherbalsolutionhome@gmail .com for more information.
ReplyDeleteI was suffering from Warts Simplex Virus, i was totally depressed due to my predicament , until i meet Dr Nana the great Traditional healer who cured me. you can also contact him now on whatsapp +2347014784614.or via email:on drnanaherbalsolutionhome@gmail .com for more information.
I am so happy, i never believe i will be this happy again in life, I was working as an air-hoster ( cabby crew ) for 3years but early this year, i loose my job because of this deadly disease called Herpes virus (HSV), I never felt sick or have any symptom, till all workers were ask to bring their doctors report, that was how i got tested and i found out that am HSV positive that make me loose my job, because it was consider as an STD and is incurable disease, i was so depress was thinking of committing suicide, till i explain to a friend of mine, who always said to me a problem share is a problem solved, that was how she directed me to Dr Isibor, that was how i contacted him and get the medication from this doctor and i got cured for real, I just went back to my work and they also carry out the test to be real sure and i was negative. Please contact this doctor if you are herpes positive diseases his email is: drisiborspellhome@gmail.com or you can call or whatsApp his mobile number on +2348107855231.
ReplyDeleteHello viewers, i want to thank Dr for his medical treatment which she used in curing my Genital herpes and am so happy that am cured completely. This is my first blessing of 2019 am so grateful to her please contact her on +1678) 723-1775 and she can also cure other manner of deadly virus email her: samherbstreatment@bk.ru or my Whatsapp number: +1 (213) 349-2159.
ReplyDeleteGod bless Dr Isibor for his marvelous work in my life, I was diagnosed of HERPES since 2017 and I was taking my medications, I wasn't satisfied i needed to get the HERPES out of my system, I searched about some possible cure for HERPES i saw a comment about Dr Isibor, how he cured HERPES DIABETES and CANCER with his herbal medicine, I contacted him and he guided me. I asked for solutions, he started the remedy for my health, he sent me the medicine through UPS SPEED POST. I took the medicine as prescribed by him and 14 days later i was cured from HERPES, Dr Isibor truly you are great, do you need his help also? Why don't you contact him through drisiborspellhome@gmail.com Or contact his number via (WHATSAPP ONLY: You can also whatsapp him via +2348107855231.
ReplyDeleteI am so happy, i never believe i will be this happy again in life, I was working as an air-hoster ( cabby crew ) for 3years but early this year, i loose my job because of this deadly disease called Herpes virus (HSV), I never felt sick or have any symptom, till all workers were ask to bring their doctors report, that was how i got tested and i found out that am HSV positive that make me loose my job, because it was consider as an STD and is incurable disease, i was so depress was thinking of committing suicide, till i explain to a friend of mine, who always said to me a problem share is a problem solved, that was how she directed me to Dr Isibor, that was how i contacted him and get the medication from this doctor and i got cured for real, I just went back to my work and they also carry out the test to be real sure and i was negative. Please contact this doctor if you are herpes positive diseases his email is: drisiborspellhome@gmail.com or you can call or whatsApp his mobile number on +2348107855231.
ReplyDeleteHello everyone, am very happy to share this little awesome testimony about Dr olu a great herbal doctor who help me enlarge my penis size.3.2 cm to 8.3 cm longer with his herbal cream mixture, my girlfriend is now so amazed with the autonomous size of my penis , if you you are also in need of help on how to enlarge your penis to become bigger and stronger I adverse you to contact Dr on his email (drolusolutionhome@gmail.com) ) you or contact on whatsapp number +2348140654426 because he is one of the best herbal doctor that i can only show you up to, if your penis is 4.2 cm and want to get it reach 9.2 cm within three weeks i Dr olu is also specialized on obey m breast and boobs enlargement i advise you to contact him for help
ReplyDeleteHello everyone, am very happy to share this little awesome testimony about Dr olu a great herbal doctor who help me enlarge my penis size.3.2 cm to 8.3 cm longer with his herbal cream mixture, my girlfriend is now so amazed with the autonomous size of my penis , if you you are also in need of help on how to enlarge your penis to become bigger and stronger I adverse you to contact Dr on his email (drolusolutionhome@gmail.com) ) you or contact on whatsapp number +2348140654426 because he is one of the best herbal doctor that i can only show you up to, if your penis is 4.2 cm and want to get it reach 9.2 cm within three weeks i Dr olu is also specialized on obey m breast and boobs enlargement i advise you to contact him for help
I'm so excited my broken Marriage has been restored my ex husband is back after he left me and our 2kids for another woman. i was so happy to met Dr. OKu how he help many people to bring there Lover back so i contact him to help me too. that was how Dr. Oku help me to bring my husband back.. A big thank to you Dr. Oku because I never thought my ex Husband will be back to me so quickly with your spell. You are the best and world greatest. if you are here and you need your Ex Lover back or your husband moved to another woman, do not cry anymore, contact this powerful spell caster now. Here’s his contact:
ReplyDeleteEmail him at: okutemple@gmail.com
You can also Call/WhatsApp:+2347053113465
I want to share a testimony of how Dr.Omohan herbal mixture cream saves me from shame and disgrace, my penis was a big problem to me as the size was really so embarrassing,and i was also having weak erection problem. I can't make love to my wife and my penis was just too small a full grown man like me having 4 inches penis and to worsen it i don't last in sex i cant even last two minutes it was really a thing of shame to me. My wife was really tired of me because my sex life was very poor,she never enjoyed sex,i was always thinking and searching for solutions everywhere until when i saw a testimony of how Dr.omohan herbal mixture cream have been helping people regarding their sex life, so i decided to give him a try and to my greatest surprise in less than one week of taking the herbs my penis grow to 8 inches i couldn't believe my eyes and as i speak now my penis is now 8 inches and i do not have week erection again. I can make love to my wife longer in bed. And my marriage is now stable,my wife now enjoy me very well in bed. can contact him dromohanherbalmedicine@gmail.com or call or what-apps him through +2348164816038. . .he also specialize on the following things
ReplyDeleteBREAST ENLARGEMENT
BREAST LIFT & REDUCTION
PENILE/PENIS ENLARGEMENT
GENERAL BODY SLIMMING
HIPS & THIGH REDUCTION
REGAIN VIRGINITY BACK
Thanks for the Enlarging my penis sir, you indeed save my marriage...I am really grateful sir,
I believed God has sent Dr Alli to help people out from this horrible virus. i was diagnosis with genital herpes for 3 years, few months ago a friend introduced me to Dr Alli, and i only took his natural treatment for 2 weeks & 3 days, i got cured permanently with his herbal treatment. Dr Alli email is Allispellhelp1@gmail.com or whats App +2348100772528
ReplyDelete
ReplyDeleteI actually was told otherwise, that the herpes can not be treated and that there is no way to make it go away once you have it. But I decided to do my own research and found out it is possible to fight it.. I got a lot of information about it here in forums and online,I got Dr Oduku contact on a group were a man from Cordova was sharing his testimony of how he cured him of herpes so I decided to give him a try I emailed him through odukuherbalremedies@gmail.com. he called me to know if I actually wanted the cured after which the cure was sent to me few days later I was surprised when the same doctor who told me that I was diagnosed of herpes told me that I was completely free from herpes you can reach him through his email odukuherbalremedies@gmail.com. or through his WhatsApp +2349064430408 if you are diagnosed and also wanna be free from this deadly virus because he is really gonna be of great help to you!!
Am so happy to share this testimony thank you Dr Oduku i never believed i will be herpes negative again despite all i passed through Dr Oduku you are truly sent by God and you can also place your order for Dr Oduku roots and herbs medicine email odukuherbalremediess@gmail.com or what-app number +2347067706774.
ReplyDeleteI has suffered for Human papillomavirus HPV) for 2years, I was given some tablets at the hospital but I refused to take it, They said I have to be on it for life so I don't want take a drugs everyday for life. No point in taking medicine everyday when u won't get cure from it and I was advice to seek for natural herbal cure, after some time I found dr onokun is the most trustful herbalist that have herbs to cure wicked symptom's,I emailed dr onokun, for 2weeks been his patient he cured my (HPV) with his herbal. I only used his natural herbs for two weeks it was 100% cure. I'm not (HPV) patient anymore. I'm happy about it i finally got cured out of this mess been in my body for 2years. I also recommend you if you're living with (HPV) or herpes symptoms i also want you to be free contact dr onokun with the email attach to my post. Dronokunherbalcure@gmail.com
ReplyDeleteAfter being in relationship with him for seven years, he broke up with me. I did everything possible to bring him back but all was in vain. I wanted him back so much because of the love I have for him and I begged him with everything, I made promises but he refused. I explained my problem to someone online and she suggested that I should rather contact a spell caster DR ILEKHOJIE that could help me cast a spell to bring him back but I am the type that never believed in spell, I had no choice than to try it. I mailed the spell caster and he told me there was no problem that everything will be okay before two days that my ex will return to me before three days, he cast the spell and surprisingly in the second day, it was around 4pm. My ex called me and I was so surprised so I answered the call and all he said was that he was so sorry for everything that happened that he wanted me to return to him that he loves me so much. I was so happy and went to him, that was how we started living together happily again. Since then, I have made promise that anybody I know that have a relationship problem, I would be of help to such person by referring him or her to the only real and powerful spell caster who helped me with my own problem and who is different from all the fake ones out there. Anybody could need the help of the spell caster, his email: gethelp05@gmail.com or his WhatsApp/Viber him on: +2348147400259
ReplyDeleteI was hurt and heart broken when a very big problem occurred in my marriage seven months ago, between me and my husband. so terrible that he took the case to court for a divorce. he said that he never wanted to stay with me again,and that he didn't love me anymore. So he packed out of the house and made me and my children passed through severe pain. I tried all my possible means to get him back,after much begging,but all to no avail.and he confirmed it that he has made his decision and he never wanted to see me again. So on one evening,as i was coming back from work,i met an old friend of mine who asked of my husband .So i explained every thing to him,so he told me that the only way i can get my husband back,is to visit a spell caster,because it has really worked for him too. So i never believed in spell,but i had no other choice,than to follow his advice. Then he gave me the Email address of the spell caster whom he visited. drajayi1990@gmail.com. So the next morning,i sent a mail to the address he gave to me,and the spell caster assured me that i will get my husband back the next day. What an amazing statement!! I never believed,so he spoke with me, and told me everything that i need to do. Then the next morning, So surprisingly, my husband who didn't call me for the past 7 months, gave me a call to inform me that he was coming back. So Amazing!! So that was how he came back that same day,with lots of love and joy,and he apologized for his mistake,and for the pain he caused me and my children. Then from that day,our relationship was now stronger than how it were before,by the help of a spell caster. So, i will advice you out there if you have any kind of problem, Please i will advice you to contact DR AJAYI, i give you 100% guarantee that he will help you solve your problems.. Email him at: drajayi1990@gmail.com OR Whatsapp : +2347084887094
ReplyDeletei already gave up on ever getting cured of HSV2 because i have try many treatment none of them work out for me i have go to different hospital they always tell me same thing there is no cure for herpes when i came across a post about Dr ohunyom, in the net from a lady called Angela i contacted him and he reassured me with him herbal medicine which i took according to the way he instructed, that how i was cured. I doubted at first because i have been to a whole lot of reputable doctors, tried a lot of medicines but none was able to cure me. so i decided to listen to him and he commenced treatment, and under two weeks i was totally fee from #Herpes. i want to say a very big thank you to DR ohunyom for what he has done in my life. feel free to leave him a message on email drohunyom@gmail.com and also WhatsApp him +2349060579973..
ReplyDeleteHe can still be able to help you with this herbs medicine:
1...ALS CURE/DIABETES CURE/EPILESY/HPV CURE/LUPUS/HEPATITIS/CANCER/GOUT
@@##HOW TO BRING BACK LOST LOVER CALL ON +27605775963 SPELLS AND BLACK MAGIC SPELLS THAT WORK IMMEDIATELY@@##
ReplyDelete,Magic Love Spells, Easy Love Spells, and Powerful Love Spells. Real Love Spells, Magic times Love Spells are used to get back your Lost Love, Break up Spells, or Love Spells or Soul Mate who is with someone else. Like Anti contact the no 1 spell caster contact divorce spells, Lovers Spells, Spell Casting Protection Spells Voodoo Dolls of love. Heal yourself now with powerful spells in the field of Love success;MAMA plays an important role in black white magic. And some of the white magic spells are Marriage Spells, Fertility Spells, Witch Craft Spells of love, **** Spells, Lust Spells, Gay Love Spells, *** Love Spells, attraction love spells, binding love spells, are in demand. Magic Love Spells for New Love or magic Love Spells to Attract new Love are very common spells. Looking for Magic Spells to attract new love or spells to attract new love. Come here where many powerful love spells are available. Love spells can be used as Protection Spells especially to Protect Marriage or Divorce as Marriage Spells. Break up Spells or Breaking Love Spells or Divorce Spells.
Call Or Whats app On +27605775963
HOW TO JOIN ILLUMINATI,SUCCESS POWERS ,PROTECTION ,+27785167256 IN South Africa,Lesotho, Namibia,Botswana, Zambia,Swaziland,Canada,Guyana, Uk,France,Germany,Spain,Poland,Switzerland,Romania,Bulgaria,Denmark,Finland,Netherlands,Norway,Sweden
ReplyDelete,Egypt,Jordan,Sudan,Tunisia,Bahrain,Iraq,Kuwait,Oman,Qatar,Saudi Arabia,UAE ,Australia,New Zealand, GHANA,Uganda,Limpopo,JORDAN, Kuwait, Saudi Arabia, Australia, Johannesburg, Lebanon,Berhrain USA, Kenya , California, Dallas, England,Spain, Austria, Vancouver, Denmark,Pretoria, Durban,Wales, France, Norway, Sweden,Capetown, Tanzania, Northern Cape, New York, Limpopo, London,Sweden, Rwanda, Oman, Qatar, Dubai, Poland, Canada, United Kingdom..
For those who are interested in making money, every good thing comes with money, comes with extra effort , All u need do is a Spiritual work and every wicked power delaying your progress wants clear and good things will come to you like, money, favor from people, open doors, business breakthrough, good job. Etc. For more info you can call +27785167256 Note: Its not a child's play, its for those who are desperate and ready to make a change in their life. Above all its 80 dollar to Join SERVICE TO HUMANITY We are seeking that special wisdom and knowledge that would set us free from the bondage to dull and dreary everyday life, while strengthening us in body, mind and spirit, and bringing us the material rewards of wealth, love, and success. The karishika Brotherhood is a true brotherhood of secret knowledge and power. membership into our fraternity is free and normally through a thorough screening. we are here to liberate those who need wealth, riches, power, prosperity, protection and success in all ramification. Agent Ben brotherhood offers all initiate members growth, wealth, fame, power, prosperity and success in all areas of heart desires. we do not demand human sacrifice, the use of any human parts or early personal death as a precondition for you to become our member. we are
not suppose to be on the internet but because of questions and comments like
Email:musumikamunilah@gmail.com
GAY/LESBIAN SPELL CASTER=LOST LOVE GAY SPELL=LOVE SPELL GAY SPELL =LOVE SPELL CASTER +27605775963
ReplyDeletestrong connection with and truly love. Lost love spells to bring back a ex-wife Did you realise how much you loved your wife after your divorce, maybe you even made the divorce request yourself. Are you regreting that your sweetheart is now your ex-wife Get my lost love spells for man to bring back a ex-wife, they work and work fast to bring back your lover and even mend things to lead to a happily ever after remarriage. Lost love spells to bring back a ex-girl friend No matter how many years you have been away from each other my lost love spells will work for you. Bring back that ex-girl friend you still love in a few days using my powerful lost love spells. Even the mistake was yours
I have used Dr Aziba herbal Medicine cure remedy for 2 WEEKS now. So far, it has cured my HERPES, eliminated my COLD SORE, improved energy and metabolism, in the morning I feel recharged and I no longer feel sleepy after a meal. As for the scent, it does smell like motor oil but I chase it down with 100% Herbal medicine from Dr Aziba. twice a day AFTER a meal. I also experienced vivid dreams for about 5 days now it’s diminishing to during the first 2 or 3 hours of sleep . contact Dr Aziba: EMAIL: priestazibasolutioncenter@gmail.com or whatsapp +2348100368288 and He also have herbs medicine to cure some popular diseases....
ReplyDeleteSome other areas of your health you may need Dr Aziba help to get cured.
{1}Diabetes
{2}HPV
{3}ALS
{4}Herpes
Contact
Dr via Email: Priestazibasolutioncenter@gmail.com
WhatsApp : +2348100368288
I'm 61 years old, I contracted hpv in 2011' I has be taking lot treatment for it and some months ago the wart stated coming out seriously, I used lot recommendation because there was lot warts around my anus and was so embarrassed. but today I'm totally happy I got the virus eliminated by using natural treatment from Dr Onokun herbal center after his treatment I got cured. all the warts went away' seriously believed Dr Onokun he have the cure for human papillomavirus because he has eliminated hpv been in my body since 2011, Dr Onokun make it possible for me. Here is Dr Onokun email to reach him: Dronokunherbalcure@gmail.com he is welled capable of curing terrible diseases.
ReplyDeleteI am here to testify about what DR. IYOHA did for me. I have been suffering from (GENITAL HERPES VIRUS) disease for the past 1 years and had constant pain and inching, especially in my private part. During the first year, I had faith in God that i would be cured someday .This disease started circulating all over my body and I have been taking treatment from my doctor, few weeks ago I came across a testimony of williams on the internet testifying about a Man called DR. IYOHA on how he cured him from 7 years HSV 2. And he also gave the email address of this man, advise anybody to contact him for help on any kind of diseases that he would be of help, so I emailed him telling him about my (HSV 2) he told me not to worry that I was going to be cured!! Well, I never doubted him I have faith he can cure me too,, DR. IYOHA prepared and sent me Healing Oil, Soap, roots and herbs which I took. In the first one week, I started experiencing changes all over me, after 3weeks of using his Roots/ Herbs, Oil and Soap, I was totally cured. no more inching , pain on me anymore as DR. IYOHA assured me. After some time I went to my doctor to do another test behold the result came out negative. So friends my advise is if you have such disease or know anyone who suffers from it or any other disease like HPV, HBV, HIV, ALS, HBP, CANCER etc. you can contact DR. IYOHA for help via email} driyohasolutiontemple@yahoo.com OR call : +1 407 337 9869. Thanks once again DR. IYOHA for making me a happy woman again ..
ReplyDeleteI Want To Appreciate Dr.OYAGU for hs great deeds, I Was Diagnosed With type 2 Herpes Virus Last year,And Was Look For Solution To Be Cured Luckily I Saw Testimonies On How Dr.OYAGU Cure Herpes Virus I Decided To Contact Dr.OYAGU I Contacted Him He Prepared A Herbal Medicine Portion And Sent It To Me,I Started The Herbal Medicine For My Health.He Gave Me Step By Step Instructions On How To Apply It, When I Applied It As Instructed, I Was Cured Of This Deadly Herpes Within 2 weeks, I Am Now Herpes Negative.My Brother And Sister I No That There Are So Many People That Have There Same Herpes Virus Please contact Dr OYAGU To Help You Too,And Help Me To Thank Dr.OYAGU For Cure Me, I’m Cured By Dr. OYAGU Herbal Medicine,His Contact Email:oyaguspellcaster@gmail.com
ReplyDeleteOr Cell Whatsapp Number +2348101755322 thank you .....
I Want To Appreciate Dr.OYAGU for hs great deeds, I Was Diagnosed With type 2 Herpes Virus Last year,And Was Look For Solution To Be Cured Luckily I Saw Testimonies On How Dr.OYAGU Cure Herpes Virus I Decided To Contact Dr.OYAGU I Contacted Him He Prepared A Herbal Medicine Portion And Sent It To Me,I Started The Herbal Medicine For My Health.He Gave Me Step By Step Instructions On How To Apply It, When I Applied It As Instructed, I Was Cured Of This Deadly Herpes Within 2 weeks, I Am Now Herpes Negative.My Brother And Sister I No That There Are So Many People That Have There Same Herpes Virus Please contact Dr OYAGU To Help You Too,And Help Me To Thank Dr.OYAGU For Cure Me, I’m Cured By Dr. OYAGU Herbal Medicine,His Contact Email:oyaguspellcaster@gmail.com
ReplyDeleteOr Cell Whatsapp Number 2348101755322 thank you .....
I Want To Appreciate Dr.OYAGU for hs great deeds, I Was Diagnosed With type 2 Herpes Virus Last year,And Was Look For Solution To Be Cured Luckily I Saw Testimonies On How Dr.OYAGU Cure Herpes Virus I Decided To Contact Dr.OYAGU I Contacted Him He Prepared A Herbal Medicine Portion And Sent It To Me,I Started The Herbal Medicine For My Health.He Gave Me Step By Step Instructions On How To Apply It, When I Applied It As Instructed, I Was Cured Of This Deadly Herpes Within 2 weeks, I Am Now Herpes Negative.My Brother And Sister I No That There Are So Many People That Have There Same Herpes Virus Please contact Dr OYAGU To Help You Too,And Help Me To Thank Dr.OYAGU For Cure Me, I’m Cured By Dr. OYAGU Herbal Medicine,His Contact Email:oyaguspellcaster@gmail.com
ReplyDeleteOr Cell Whatsapp Number 2348101755322 thank you .....
ReplyDeletei couldn't believe that i would ever be re-unite with my ex-lover, i was so traumatize staying all alone with no body to stay by me and to be with me, but i was so lucky one certain day to meet this powerful spell caster Dr Akhere,after telling him about my situation he did everything humanly possible to see that my lover come back to me,indeed after casting the spell my ex-lover came back to me less than 48 hours,my ex-lover came back begging me that he will never leave me again,3 months later we got engaged and married,if you are having this same situation just contact Dr Akhere on his email: AKHERETEMPLE@gmail.com thanks very much sir for restoring my ex-lover back to me,his email: AKHERETEMPLE@gmail.com or call/whatsapp:+2349057261346
hindi ako makapaniwala na kailanman ay muling makiisa ako sa aking kasintahan, labis akong na-trauma sa pananatiling nag-iisa na walang katawan na manatili sa akin at makakasama ko, ngunit napakasuwerte ako sa isang tiyak na araw upang matugunan ito malakas na spell caster na si Dr Akhere, matapos sabihin sa kanya ang tungkol sa aking sitwasyon ginawa niya ang lahat ng makataong posible upang makita na ang aking kasintahan ay bumalik sa akin, sa katunayan matapos na ihagis ang spell ang aking dating kasintahan ay bumalik sa akin ng mas mababa sa 48 oras, dumating ang dating kasintahan ko. bumalik sa pagmamakaawa sa akin na hindi na niya ako pababayaan, 3 buwan mamaya kami ay nakipag-ugnay at nag-asawa, kung nagkakaroon ka ng parehong sitwasyong ito makipag-ugnay lamang kay Dr Akhere sa kanyang email: AKHERETEMPLE@gmail.com maraming salamat sa sir sa pagpapanumbalik ng aking dating kasintahan bumalik sa akin, ang kanyang email: AKHERETEMPLE@gmail.com o tumawag / whatsapp: +2349057261346
Hello everyone I was diagnosed of HERPES Virus in year 2000 and I have tried all I can to get cured but all to no avail, until i saw a post in a health forum about a herbalist man who prepare herbal medication to cure all kind of diseases including HERPES virus, at first i doubted if it was real but decided to give it a try, when i contact this herbalist via his email and he prepared a HERPES herbal cure and sent it to me via UPS delivery service, when i received this herbal cure, he gave me step and Instructions on how to apply it, when i applied it as instructed, i was totally cured of this disease within 17-21 days of usage, I am now free from this horrible illness disease called herpes, All thanks to Dr Iyoha for saving my life please if you are herpes simplex virus patient, contact him and I am sure you will get cured, contact him via: driyohasolutiontemple@yahoo.com or call : +1 407 337 9869. He also have the herb to cure: – ALS/HPV – LUPUS – HIV&AIDS, – OVARIAN CYST, – CANCER – HSV-2/KIDNEY-FAILURE -CYMBALTA – ARTHRITIS.
ReplyDeleteHello, everyone! I,m here to explore blogs and forum about the wonderful and most safe cure for (Herpes Virus).I was positive to the deadly virus called herpes and i lost hope because i was out casted and rejected even by my closet friends.i searched on-line to know and enquirer about cure for Herpes and i read someone testimony i know that nature has the power to heal everything.i contacted him to knoon how he was cured from Herpes so i decided to contact the same herbalist becausew how he can help me and he told me never to worry that he will heal me with the natural herbs from God!after 2 days of contacting him, he told me that the cure has been ready and he sent it to me via DHL and it got to me after 3 days!i used the med as he instructed me (MORNING and EVENING) and i was cured!its really like a dream but i am so happy!thats the reason i decided to also add more comment of Him so that more can be saved just like me!and if you need his help, you can email him on (drakuzasolutioncenter@gmail.com) : whatsapp+2348072437003 I,m neme amber and you can get in touch with Contact him for help at Herpes virus HIV/AIDS CANCER COPD BRAIN TUMOR All kind of virus and disease.....
ReplyDeleteHello, everyone! I,m here to explore blogs and forum about the wonderful and most safe cure for (Herpes Virus).I was positive to the deadly virus called herpes and i lost hope because i was out casted and rejected even by my closet friends.i searched on-line to know and enquirer about cure for Herpes and i read someone testimony i know that nature has the power to heal everything.i contacted him to knoon how he was cured from Herpes so i decided to contact the same herbalist becausew how he can help me and he told me never to worry that he will heal me with the natural herbs from God!after 2 days of contacting him, he told me that the cure has been ready and he sent it to me via DHL and it got to me after 3 days!i used the med as he instructed me (MORNING and EVENING) and i was cured!its really like a dream but i am so happy!thats the reason i decided to also add more comment of Him so that more can be saved just like me!and if you need his help, you can email him on (drakuzasolutioncenter@gmail.com) : whatsapp+2348072437003 I,m neme amber and you can get in touch with Contact him for help at Herpes virus HIV/AIDS CANCER COPD BRAIN TUMOR All kind of virus and disease.....
ReplyDeleteHappiness is all i see now I never thought that I will live on
ReplyDeleteearth before the year runs out. I have been suffering from a
deadly disease (Herpes) for the past 3 years now; I had spent
a lot of money going from one places to another, from
churches to churches, hospitals have been my home every day
residence. Constant checks up have been my hobby not until
this faithful day, I was searching through the internet, I saw a
testimony on how pp him +2348154637647 Dr Lucky, helped
someone in curing his Herpes disease, quickly I copied his
email which is (drluckyherbalcure@gmail.com) just to give
him a test I spoke to him, he asked me to do some certain
things which I did, he told me that he is going to provide the
herbal cure to me, which he did, then he asked me to go for
medical checkup after some days after using the herbal cure, I
was free from the deadly disease, he only asked me to post
the testimony through the whole world, faithfully am doing it
now, please brothers and sisters, he is great, I owe him in
return. if you are having a similar problem just email him on
(drluckyherbalcure@gmail.com) or Call him or WhatsApp him
+2348154637647
God bless Dr ojoka for his marvelous work in my life, I was diagnosed of HERPES since 2011 and I was taking my medications, I wasn't satisfied i needed to get the HERPES out of my system, I searched about some possible cure for HERPES i saw a comment about Dr ojoka how he cured HERPES with his herbal medicine, I contacted him and he guided me. I asked for solutions, he started the remedy for my health, he sent me the herbal medicine through UPS SPEED POST. I took the medicine as prescribed by him in 2 weeks and 3 days later i was cured from HERPES, Dr ojoka truly you are great, do you need his help also? Why don't you contact him through email:{dr.ojokarootandherbal@gmail.com or whats app/call +2348144172934} check his blog: https://perfectherbalcure.blogspot.com/ check his blog: https://penisherbalenlarge.blogspot.com/ FB page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/
ReplyDeleteGod bless Dr ojoka for his marvelous work in my life, I was diagnosed of HERPES since 2011 and I was taking my medications, I wasn't satisfied i needed to get the HERPES out of my system, I searched about some possible cure for HERPES i saw a comment about Dr ojoka how he cured HERPES with his herbal medicine, I contacted him and he guided me. I asked for solutions, he started the remedy for my health, he sent me the herbal medicine through UPS SPEED POST. I took the medicine as prescribed by him in 2 weeks and 3 days later i was cured from HERPES, Dr ojoka truly you are great, do you need his help also? Why don't you contact him through email:{dr.ojokarootandherbal@gmail.com or whats app/call +2348144172934} check his blog: https://perfectherbalcure.blogspot.com/ check his blog: https://penisherbalenlarge.blogspot.com/ FB page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/
Hello everyone,am from USA, I’m here to testify to the general public on how my mother DIABETES INFECTION was cure by Dr oriane, Thanks goes to Dr. oriane who finally save my son life with his herbal medication at low cost,for over 7 years now my mother has been suffering from DIABETES on which we had gone to various hospital for treatment but to no avail until a friend recommend Dr oriane herbal remedy to me which i decided to contact,he advice and assure me that my mother will be cured with his herbal medicine ,i listen to the advice and instruction given to me by Dr oriane ,he prepare and send me the medication which i use on my mother health ,within 30 days my mother was totally cured from DIABETES and today am very happy and May God continue to support you for the good work you are doing Dr oriane, i decided to share This Testimony to the general public for those that need his help. if you are out their suffering in pains looking for solution for any spiritual problem or infections of any kinds.Contact him Via this Email : droriane6@gmail.com, He is out there to help you out and put a smile on your face again. also he can cure any kinds of deadly disease, Herpes, Stroke, Herbs and Cancer. Truly 100% sure treatment for just 30 days as instructed on how to use his herbal medication all the pains are gone,
ReplyDeleteHello everyone,am from USA, I’m here to testify to the general public on how my mother DIABETES INFECTION was cure by Dr oriane, Thanks goes to Dr. oriane who finally save my son life with his herbal medication at low cost,for over 7 years now my mother has been suffering from DIABETES on which we had gone to various hospital for treatment but to no avail until a friend recommend Dr oriane herbal remedy to me which i decided to contact,he advice and assure me that my mother will be cured with his herbal medicine ,i listen to the advice and instruction given to me by Dr oriane ,he prepare and send me the medication which i use on my mother health ,within 30 days my mother was totally cured from DIABETES and today am very happy and May God continue to support you for the good work you are doing Dr oriane, i decided to share This Testimony to the general public for those that need his help. if you are out their suffering in pains looking for solution for any spiritual problem or infections of any kinds.Contact him Via this Email : droriane6@gmail.com, He is out there to help you out and put a smile on your face again. also he can cure any kinds of deadly disease, Herpes, Stroke, Herbs and Cancer. Truly 100% sure treatment for just 30 days as instructed on how to use his herbal medication all the pains are gone,
ReplyDeleteHELLO EVERYONE.. FEW MUNINETS TO REDY THIS INFOR ON HERPES CURE 2018..
ReplyDelete2017 MY MOTHER WAS DIAGNOSED OF HERPES/ KNOWN AS GENITAL WARTS ,I SPENT A LOT OF MONEY ON HER MEDICATION TILL A POINT I EVEN LOST HOPE,BECAUSE MY MOTHER WAS GRADUALLY DYING AND LOST HER MEMORY TOO, I WAS SO DESPERATE TO GET MY MOTHER BACK TO NORMAL, ONE DAY MY UNCLE WHO LIVES IN LONDON UNITED KINGDOM TOLD ME ABOUT DR OLIHA ,WHO HELPED HIM GET RID OF HERPES /GENITAL WART WITH HERBAL MEDICINE AND HIS HERBAL SOAP ,I WAS SO SHOCKED WHEN HE TOLD ME ABOUT THIS ,ALTHOUGH I NEVER BELIEVE IN HERB BUT, I KEEP TO BELIEVE BECAUSE MY UNCLE CAN'T TELL ME LIES WHEN IT COMES TO HEALTH CONDITION I CONTACTED DR OLIHA VIA HIS EMAIL; OLIHA.MIRACLEMEDICINE@GMAIL.COM , YOU CAN TALK TO HIM VIA CALL OR WHATSAPP MESSENGER ON +2349038382931 , HE REPLIED AND ASK ME TO SEND MY HOME ADDRESS AND MY MOTHER'S DETAIL AND THEN I PURCHASED THE HERBAL MEDICINE,SENT ME THE HERBAL MEDICINE THROUGH COURIER SERVICE, WHEN I RECEIVED THIS HERBAL MEDICINE USED IT FOR 2 WEEKS, AND 4 DAYS OF USAGE THE WARTS FELL OFF, MY MOTHER I NOW TOTALLY CURED AND MY MOTHER IS LIVING FREE AND HAPPY AGAIN. YOU CAN TALK TO DR VIA HIS MOBILE NUMBER OR WHATS APP HIM ON +2349038382931.ALL THANKS TO DOCTOR DR OLIHA
HOW I GOT RID OF MY HERPES Virus WITH THE HELP OF DR AKERE. Greetings to the general public, i want to inform the general public how i was cured of HERPES Simplex Virus by a Doctor called Dr AKERE. i visited different hospital but they gave me list of drugs like Famvir, Zovirax, and is Valtrex which is very expensive to treat the symptoms and never cured me. I was browsing through the Internet searching for remedy on HERPES and i saw comment of people talking about how Doctor Dr AKERE cured them. when i contacted him he gave me hope and send a Herbal medicine to me that i took as instructed and it seriously worked for me, i am a free person now without problem, my HERPES result came out negative. I pray for you Dr AKERE God will give you everlasting life, you shall not die before your time for being a sincere and great man. Am so happy, you can also contact him if you have any problem like HIV/AID, CANCER,HERPATITIS,DIABETES,HPV,INFECTION ETC. Reach him on his Email Address at=== drakereherbalcure@gmail.com or you can contact him on WHATSAPP FOR EASY ACCESS at +2348070670337
ReplyDeleteWEBSITE: https://drakereherbalcure.wixsite.com/website-2
BLOG:
https://drakereremedy.blogspot.com/
I am a new user of this site so here i saw multiple articles and posts posted by this site,I curious more interest in some of them hope you will give more information on this topics in your next articles. https://www.germany-medicare.de/product/clomid-clomifen/
ReplyDeleteI Want To Appreciate Dr.OYAGU for hs great deeds, I Was Diagnosed With type 2 Herpes Virus Last year,And Was Looking For Solution To Be Cured Luckily I Saw Testimonies On How Dr.OYAGU Cure Herpes Virus I Decided To Contact Dr.OYAGU I Contacted Him He Prepared A Herbal Medicine Portion And Sent It To Me,I Started The Herbal Medicine For My Health.He Gave Me Step By Step Instructions On How To Apply It, When I Applied It As Instructed, I Was Cured Of This Deadly Herpes Within 2 weeks, I Am Now Herpes Negative.My Brother And Sister I No That There Are So Many People That Have There Same Herpes Virus Please contact Dr OYAGU To Help You Too,And Help Me To Thank Dr.OYAGU For Cure Me, I’m Cured By Dr. OYAGU Herbal Medicine,His Contact Email:oyaguherbalhome@gmail.com
ReplyDeleteOr Cell Whatsapp Number +2348101755322 thank you
Can't still believe that i got cured from Genital Herpes through herbal treatment from Dr LUCKY who I met through the internet, I actually couldn't believe it at first because it sounded impossible to me knowing how far I have gone just to get rid of it. Dr LUCKY send me his medicine which I took as instructed and here I am living a happy life once again, a big thanks to Dr LUCKY , I am sure there are many herbal doctors out there but Dr LUCKY did it for me, contact him on Email him; { drluckyherbalcure@gmail.com }
ReplyDeleteHello everybody I am here to show appreciation to Dr Robinson the great herbal doctor that help me got rid of my herpes virus which i contacted, And after so many search looking for medical cure to my illness which no medical doctor had cure to with no choice I was forced to share my pain to a friend who had bell's palsy and she directed me to this amazing herbal doctor with great powers who got her total cure and she assured me that Dr Robinson will help me get total rid of my herpes virus and f with the help of his herbal herbal cure and so surprising after taking his herbal treatment i was totally cure and for 2 months now i have not experienced any symptoms i am so happy today to tell you all my story though it's cost me some money but I guarantee you he is one man you can contact for help and he will help you with his great powers. So if you have been looking for cure to whatever illness you are suffering from contact Dr Robinson by visiting his drrobinsonspellhome@gmail.com or call and you will be glad you came to him for help for I strongly believe Dr Robinson will never fail in his herbal solutions .
ReplyDelete
ReplyDeleteThank you Dr Williams for what you have done for me am so greatful my lover is back to me and we are now living happily together. Dr Williams love spell is very powerful and effective and it does not have any side effect as he promised I decided to give Dr Williams a try when my lover left me for another he helped me to cast a love spell on my lover that brought him back to me what makes me excited the most is that my lover did not even know he is under a spell if you are passing through relationship love problem I advice you to contact Dr Williams to get your problem solve. It's very hard to loose a love one and I know how it feels so do not let somebody take away your lover from you. Contact Dr Williams today and get your problem solve and if you need his help below here is his email address.
drwilliams533@gmail. com or you can also reach him on his WhatsApp +2348136785562
Traditional Herbal doctor Sayo Cure HSV-2 Disease
ReplyDeleteI want to thank Dr Sayo for helping me with his Herbal Medicine that cure my HSV-2 Disease completely.i have been living with HSV-2 Disease for the past 3yrs. i was so happy when i found DrSayo blogs: how Dr Sayo help people in the world.to cure HSV, with Herbal Medicine he Cured My HSV -2 Negative after using the Herbal Medicine Dr Sayo sent to me. I give thanks to God and healed completely. you are the best Traditional Herbal doctor in the world Dr Sayo, Contact Doctor Sayo Email sayoherbalhealer@gmail. com OR Contact Sayo by check FB Page https://www.facebook.com/Sayo-Herbal-Healer-100145798345000/ check website; http://sayoherbalhealer.website2.me/ check blog: https://sayoherbalhealer.blogspot.com/
Hello everyone out there, I am here to give my testimony about a herbalist doctor who helped me. I was infected with Hepatitis b (3) years now, I went to many hospitals to heal myself but there was no solution, so I was thinking how I can get a solution so that my body can be well. One day I was searching on internet I found a lady testifying on how Dr edu cure her Hepatitis b and contact him and He told me everything I had to do and also gave me instructions to take, which I followed correctly. Before I knew what was happening after two weeks, the Hepatitis b that was in my body disappeared. therefore, if you also have a broken heart and need help, you can also email him dredu933@gmail.com or whatsapp him number: +2348121760698Contact him today and you will have a testimony ... Good luck also have cure for:HERPES 1/2Diabetes 1/2ALS (Lou Gehrig's disease)Hepatitis And B Pregnancy problem thanks you so much.
ReplyDeleteI Never believed i was ever going to be HSV FREE again, DR.Ehiaguna has given me reasons to be happy, i was HSV positive for 2 years and all the means and medicine i tried for treatment was not helpful to me, but when i came on the Internet i saw great testimony about DR.Ehiaguna on how he was able to cure someone from HSV 2, this person said great things about this man, and advice we contact him for any Disease problem that DR.Ehiaguna can be of help, well i decided to give him a try, he requested for my information which i sent to him, and he told me he was going to prepare for me a healing portion, which he wanted me to take for days, and after which i should go back to the hospital for check up, well after taking all the treatment sent to me by DR.Ehiahuna , i went back to the Hospital for check up, and now i have been confirmed HSV NEGATIVE, friends you can reach DR.Ehiaguna on any treatment of any Disease cos i saw many testimony of different disease like, HEPATITIS,HIV AIDS,EPILEPSY, CANCER,CFS he is the one only one i can show you all up to, reach him on drehiaguna@gmail.com or whatsApp him now +2348073908953. quick contact him for help and you can just quickly drop your number on your first mail as i did for easily conversation
ReplyDelete
ReplyDeleteHELLO EVERYONE
Dr Wonder has really be so wonderful to me i will always testify of his greatness in my life who could have believe Dr Wonder herbal herbs cured me from my herpes virus which i was having for the past 4 year,i came across Dr.Wonderspellhome@yahoo.com on a page of how to treat herpes virus and i saw a lot of testimony of how great Dr Wonder has helped them in there various problems and situations and his mobile numbers were also on each testimony +2349059484309
to contact him, because i needed help to get rid of my herpes virus i wrote him on whats app and told him what i was passing through!dear friends all Dr Wonders ask me was not much to make sure am cured from my herpes virus, remember health is wealth, he sent me a herbal remedy for the cure of my herpes virus i was so much happy because after drinking the herbs as i was instructed by him i went to the hospital for checkup to my surprise my herpes result came out negative i will recommand Dr.Wonderspellhome@yahoo.com to anyone who need his help you can also whatsapp him +2349059484309
...Thanks Dr
NATURAL HERBS AND VEGANS CURE FOR Herpes 1&2 THAT WORK FAST WITHIN 14 DAYS WITH DR ELINFOH HERBAL MEDICINE, I saw so many testimonies about Dr ELINFOH a great HERBAL DOCTOR that will help you CURE and give you the rightful health to live a joyful life, i didn't believe it at first, but as the pain got worsen and my life was at risk after using lots of ARV drugs from the hospital and no changes so i decided to give a try to Dr Elinfoh I contacted him also and told him i want a cure for Herpes , he gave me advice on what i must do and he deliver it to me in my home address i gave him and i got the medicine which i use according to his instruction, and today i must say I am so grateful to this man Dr Elinfoh for curing me from Herpes and for restoring me back to my normal health and a sound life and i am making this known to every one out there who have been trying all day to be cured from Herpes or any sickness should not waste more time just contact him with his details below, believe me this is the only way to get cured from Hepres , this is the real solution we all have been searching for. Do not waste more time contacting him today for you can also leave a sound and happy life. contact info below. Email: (drelinfohherbalhome@gmail.com ) or WhatsApp or Call him on: +2348110492072. and you can as well contact him on other issues you are having like*HEPATITIS B*DIABETICS*CANCER*ALS*HERPES*BODY REDUCTION*BREAST ENLARGEMENT*FIBROD*CANCER*PREGNANCY MISCARRIAGE*Relationship issues etc.. ** Neoplasms*Cardiovascular Disease (CVD)
ReplyDelete*Chronic Respiratory Diseases
* Mental and Behavioral Disorders
*Alzheimer’s Disease
Email: (drelinfohherbalhome@gmail.com).
I never thought that there will be a miracle on the internet until i came in contact with Priest Babaka, finally i made it with his help, with his spiritual power which was recommended by a lady in baby center he help before, i never believe it was real until i confirm it now because i have tried so many things to make sure i get pregnant but no luck, immediately i contact him. he did some spiritual prayers and send me a medicine which i took, i got pregnant three weeks after, and i now have a son to show. thank you so much priest and i recommend Priest Babaka for everyone out there that is willing to have a child of her own out there. contact him for help too he is real and powerful, i have confirmed it, contact him on email: babaka.wolf@gmail.com or Facebook at priest.babaka
ReplyDeleteI never thought that there will be a miracle on the internet until i came in contact with Priest Babaka, finally i made it with his help, with his spiritual power which was recommended by a lady in baby center he help before, i never believe it was real until i confirm it now because i have tried so many things to make sure i get pregnant but no luck, immediately i contact him. he did some spiritual prayers and send me a medicine which i took, i got pregnant three weeks after, and i now have a son to show. thank you so much priest and i recommend Priest Babaka for everyone out there that is willing to have a child of her own out there. contact him for help too he is real and powerful, i have confirmed it, contact him on email: babaka.wolf@gmail.com or Facebook at priest.babaka
Unbelievable..... Is like a dream but is real, @dr_ahonsi the real cure to all diseases, @dr_ahonsi cure my (HERPES SIMPLEX VIRUS) that was in my body for over 6 years, The story start like this, I contacted Herpes from my ex-husband, It was hell, i was in search of a cure for years, until a friend introduce me to @dr_ahonsi I thought he was a scam, but i gave him a try, he told me what to purchase to prepare the Herbal Medicine, so i buy all the required items, before 2 days he prepare the medicine and send it to me, i take the medicine for 3 weeks then i went back for check up, the Doctor was even surprise to tell me that am cured, Thank you @dr_ahonsi for bringing happiness to my life, you can also contact him today if you have any disease, Email him :(drahonsi517@gmail.com) or whatsapp him:+2349046402730 for your complete cure
ReplyDeleteHerpes Cure Testimony
ReplyDeleteI was diagnose with Genital Herpes for the past 2 years and I have been searching for cure. I have several outbreaks on my back and it really affected me morally, I read a testimony on this platform of a lady from Nevada who was cured from Diabetes with doctor Nelson Saliu Herbal medicine including the doctor official email address. I contacted the doctor through his email, after much discussion and few questions he prepared the Herbal medicine and asked for my address which I received the Herbal medicine 3 days later and with his prescription I drank the Herbal medicine for 21 days. After concluding the herbal medicine I went for test and my IgG result was confirmed Negative with no trace of the virus on my blood. Contact doctor Nelson and be cured his email;
drnelsonsaliu10@gmail.com or send him a whatsapp text +2348111067336
He has Herbal cure for Diabetes, Hepatitis, Heart diseases.
Hypertension, Alzheimer's disease, cancer, Strokes and Liver disease.
I have been suffering from Herpes for the past 3 years and 8 months, and ever since then i have been taking series of treatment but there was no improvement until i came across testimonies of Dr Ogedegbe on how he has been curing different people from different diseases all over the world, then i contacted him as well. After our conversation he sent me the medicine which i took according to his instructions. When i was done taking the herbal medicine i went for a medical checkup and to my greatest surprise i was cured from Herpes. My heart is so filled with joy. If you are suffering from Herpes or any other disease you can contact Dr Ogedegbe today on this Email address: dr.ogedegbe6@gmail.com or WhatsApp him on this Number +2348109374702 .... he also has the cure for the following..............
ReplyDeleteHPV......ALS....COPD......HIV/AIDS.....HEPATITIS A,B,C.........
HERPES CURE....THIS IS REAL ! TAKE TIME TO READ
ReplyDeleteTESTIMONY OF HOW I GOT CURED FROM GENITAL HERPES SIMPLEX VIRUS WITHIN 14DAYS[with Herbal Traditional medicine]
Hello Everyone out there, I'm Toshia Cook, from Texas USA. I am here to give my testimony about a Herbalist called Dr SAYO, who helped me in my life. I was infected with HERPES SIMPLEX VIRUS 2 in 2010, i went to many hospitals for cure but there was no solution, so I was thinking how can I get a solution out so that my body can be okay. One day I was at the river side thinking where I could get solution. so a lady walked to me asking me why I was so sad and i opened up all to her telling her my problem, she told me that she could help me out, she introduced me to a doctor who uses herbal medication to cure HERPES SIMPLEX VIRUS 2 and gave me his email, so i mailed him. He told me all the things I needed to do and also gave me instructions, which I followed properly. Before I knew what is happening after two weeks the HERPES SIMPLEX VIRUS that was in my body got vanished . so if you are also heart broken and also need a help, you can also email him via: (sayoherbalhealer@gmail .com)
He also have a herbal cure for HIV,
CANCER,
ASTHMA,
IMPOTENCE,
BARENESS/INFERTILITY ...
CURE YOUR SELF FROM THAT ILLNESS TODAY,DON'T SPREAD YOUR INFECTION OR DISEASES TO OTHER PEOPLE.
Contact him today and you will have a testimony...Good luck
Alpha Brain Memory Enhancement System is the answer to all those people who have been looking for a cure for their memory loss.
ReplyDeleteGreetings everyone.
ReplyDeletei am very happy to share this little awesome testimony about Dr OKISIN a great herbal doctor who help me enlarge my penis size.3.2 cm to 8.3 cm longer with his herbal cream mixture, my girlfriend is now so amazed with the autonomous size of my penis , if you you are also in need of help on how to enlarge your penis to become bigger and stronger I advIce you to contact Dr OKISIN on his email (dr.okisinsolutiontemple@gmail.com) ) or contact him on whatsapp number +2348109374702 because he is one of the best herbal doctor that i can only show you up to, if your penis is 4.2 cm and want to get it reach 9.2 cm within three weeks i recommend Dr OKISIN he is also specialized on breast and boobs enlargement i will advise you to contact him for help DR OKISIN whatsapp number +2348109374702...........
My life was destroyed when my husband sent me packing, after 13 years we have been together. I was lost and helpless after trying so many ways to my husband back to me. One day at work, I was distracted, not knowing that my boss called me, so he sat and asked me what it was all about, I told him and he smiled and said it was no problem. I never understood what he meant by it was no problem getting back my husband, he said he used a spell to get back his wife when she left him for another man, and now they are together till date and initially I was shocked hearing something from my boss. He gave me an email address of the LORD NOBEL which helped him get his wife back, I never believed that this would work, but I had no choice coming into contact with the sayings that I get done, and he asked for my information and that my husband was able to propose to throw him the spell and I sent him the details, but after two days, my mother called me that my husband was pleading that he wants me back, I never believed, because it was just a dream and I had to rush off to my mother's place and to my greatest surprise, was kneeling my husband beg me for forgiveness that he wants me and the child back home, when I gave LORD NOBEL a conversation regarding sudden change of my husband and he made clear to me that my husband will love me until the end of the world, that he will never leave for another woman. Now me and my husband is back together and started doing funny things he has not done before, he makes me happy and do what it is supposed to do as a man without nagging. Please if you need help of any kind need, please contact LORD NOBEL for help. His email is allmightbazulartemple@gmail.com or https://allmightbazulartem.wixsite.com/ whatsap +2347051758952
ReplyDeleteAm Laura Mildred by name, i was diagnosed with Herpes 4 years ago i lived in pain with the knowledge that i wasn't going to ever be well again i contacted so many herbal doctors on this issue and wasted a large sum of money but my condition never got better i was determined to get my life back so one day i saw Mr. Morrison Hansen post on how Dr. Emu saved him from Herpes with herbal medicine i contacted Dr. Emu on his Email: Emutemple@gmail.com we spoke on the issue i told him all that i went through and he told me not to worry that everything will be fine again so he prepared the medicine and send it to me and told me how to use it, after 14 days of usage I went to see the doctor for test,then the result was negative, am the happiest woman on earth now thanks to Dr. Emu God bless you. Email him at: Emutemple@gmail.com Call or Whats-app him: +2347012841542
ReplyDeleteI have been suffering from Herpes for the past 3 years and 8 months, and ever since then i have been taking series of treatment but there was no improvement until i came across testimonies of Dr Sayo on how he has been curing different people from different diseases all over the world, then i contacted him as well. After our conversation he sent me the medicine which i took according to his instructions. When i was done taking the herbal medicine i went for a medical checkup and to my greatest surprise i was cured from Herpes. My heart is so filled with joy. If you are suffering from Herpes or any other disease you can contact Dr Sayo today on this Email address: (sayoherbalhealer@gmail.com) or WhatsApp him on this Tell.Number +2349012175679 Visit his FB page:
ReplyDeletehttps://www.facebook.com/Sayo-Herbal-Healer-100145798345000/ visit website: http://sayoherbalhealinghome.com/
Am here to appreciate Dr Ihibor for using his herbal medicine to cure my Herpes virus. since 3 years now I have been living with this virus and it has been giving me challenges, I was so perplexed cause i have been taking several drugs to be cured but all of my effort was in vain, one morning i was browsing through the internet then i saw several testimonies about Dr. Ihibor curing people from Herpes virus and many other illnesses in the body systems, and immediately i contacted Dr Ihibor on his email: drihiborherbalhome@gmail.com, I told him about my troubles and he told me that i must be cured, he gave me some instructions and which i rightly followed. so he prepared a herbal medicine for me and sent it to me which i used for 2 weeks and everything was like a dream to me and my Herpes virus was totally gone, why don’t you contact him today and be free from your diseases because he is very good and honest Doctor. contact him via email; drihiborherbalhome@gmail.com / his mobile whats-app +2349050141449....One thing i love most about Dr Ihibor is honestly and he is very polite with his patience, so everything he told me was what he did, and his herbal medicine are very affordable to get, Dr Ihibor god will over bless you.
ReplyDeleteI have been suffering from Herpes for the past 3 years and 8 months, and ever since then i have been taking series of treatment but there was no improvement until i came across testimonies of Dr Ihibor on how he has been curing different people from different diseases all over the world, then i contacted him as well. After our conversation he sent me the medicine which i took according to his instructions. When i was done taking the herbal medicine i went for a medical checkup and to my greatest surprise i was cured from Herpes. My heart is so filled with joy. If you are suffering from Herpes or any other disease you can contact Dr Dr Ihibor today on his Whats-app him on +2349050141449 or Email: drihiborherbalhome@.com Dr. Dr Ihibor also cures: 1. HIV / AIDS 2. HERPES 1/2 3. CANCER 4. ALS (Lou Gehrig's disease) 5. Hepatitis B 6. chronic pancreatic 7. emphysema 8. COPD (chronic obstructive pulmonary disease, etc !!
ReplyDeleteThis is the type of information I’ve long been trying to find. Thank you for writing this information. viagra generico online
ReplyDeleteInvesting online has been a main source of income,that's why knowledge plays a very important role in humanity,you don't need to over work yourself for money.All you need is the right information,and you could build your own wealth from the comfort of your home!Binary trading is dependent on timely signals,assets or controlled strategies which when mastered increases chance of winning up to 90%-100% with trading. It’s possible to earn $10,000 to $20,000 trading weekly-monthly,just file a complaint with Robert,I had almost given up on everything about binary trading and ever getting my lost funds back,till i met with him,with his help now i have my lost funds back to my bank account and I can now trade successfully with his profitable strategies and software!! Email: Robertseaman939@gmail.com or Fb.me/investandmakemoney1 or whatsApp: +44 7466 770724
ReplyDeleteI Want To Appreciate Dr. momoh for his great deeds, I Was Diagnosed With Herpes Virus Last 2018, And i Was Looking For Solution To Be Cured Luckily I Saw Testimonies On How Dr. momoh Cure Herpes Virus I Decided To Contact Dr. momoh I Contacted Him He Prepared A Herbal Medicine Portion And Sent It To Me,I Started The Herbal Medicine For My Health.He Gave Me Step By Step Instructions On How To Apply It, When I Applied It As Instructed, I Was Cured Of This Deadly Herpes Within 2 weeks, I Am Now Herpes Negative.My Brother And Sister I Know That There Are So Many People That Have The Same Herpes Virus Please contact Dr momoh To Help You Too, And Help Me To Thank Dr. momoh For Curing Me, I’m Cured By Dr. momoh Herbal Medicine, His Contact Email:(dr.momoh9@gmail.com) Or Cell Whatsapp Number +234 708 372 4098 thank you so much DR.momoh. He can also cure other diseases/infections
ReplyDeleteHello! I'm very excited to inform everyone that I'm completely cured from my HSV 1&2 recently. I have used Oregano oil, Coconut oil, Acyclovir, Valacyclovir, Famciclovir, and some other products and it's really help during my outbreaks but I totally got cured! from my HSV with a strong and active herbal medicine ordered from a powerful Traditional Healer and it completely fought the virus from my nervous system and I was tested negative after 12 days of using the herbal medicine. I'm here to let you' all know that herpes virus has a complete cure, I got rid of mine with the help of DR ODUDU a traditional Healer from west African and his herbal exploit. Contact him via email: dr.oduduherbalhome@gmail.com or +2348101571054 and his website https://dr-odudu-herbal-home.webnode.com
ReplyDeleteHello! I'm very excited to inform everyone that I'm completely cured from my HSV 1&2 recently. I have used Oregano oil, Coconut oil, Acyclovir, Valacyclovir, Famciclovir, and some other products and it's really help during my outbreaks but I totally got cured! from my HSV with a strong and active herbal medicine ordered from a powerful Traditional Healer and it completely fought the virus from my nervous system and I was tested negative after 12 days of using the herbal medicine. I'm here to let you' all know that herpes virus has a complete cure, I got rid of mine with the help of DR ODUDU a traditional Healer from west African and his herbal exploit. Contact him via email: dr.oduduherbalhome@gmail.com or +2348101571054 and his website https://dr-odudu-herbal-home.webnode.com
ReplyDeletemy partner and I have been trying for a baby for over two years now, We were going to a fertility clinic for about 5 months before somebody at baby center told us to contact this spell caster who is so powerful, We contacted him at this email; babaka.wolf@gmail.com or Facebook at priest.babaka , for him to help us, then we told him our problem, he told us that we will conceive once we follow his instructions ,but after two years of trying we were at a point where we were willing to try anything. And I'm glad we came to Priest Babaka, Because his pregnancy spell cast and herbal remedy help us, and I honestly believe him, and his gods really helped us as well, I am thankful for all he has done. contact him via email: babaka.wolf@gmail.com or Facebook at priest.babaka if you are trying to have a baby or want your lover back. he has powers to do it, he has done mine
ReplyDeletemy partner and I have been trying for a baby for over two years now, We were going to a fertility clinic for about 5 months before somebody at baby center told us to contact this spell caster who is so powerful, We contacted him at this email; babaka.wolf@gmail.com or Facebook at priest.babaka , for him to help us, then we told him our problem, he told us that we will conceive once we follow his instructions ,but after two years of trying we were at a point where we were willing to try anything. And I'm glad we came to Priest Babaka, Because his pregnancy spell cast and herbal remedy help us, and I honestly believe him, and his gods really helped us as well, I am thankful for all he has done. contact him via email: babaka.wolf@gmail.com or Facebook at priest.babaka if you are trying to have a baby or want your lover back. he has powers to do it, he has done mine
I want to share this wonderful testimony to the Good people all over the world on how I was able to Enlarge my Penis by Dr. Aziba. I was living a shameful life from my young age, just last month as I was browsing on the internet about Penis size and Enlargement Products, I saw a testimony of a Man called Tim James testifying of how he was able to get his penis Enlarged by Dr. Aziba and I decided to also Email Dr. Aziba for my small penis size and he quickly respond to me and gave me the normal instructions which i did and then he shipped the product to me here in the united state which i received in just 3 working days and today i am very happy because i started seeing positive changes in my penis size in just 7 days of use. Dr Aziba herbal product is the best recommended for you and to whom ever suffering from this shame or having any other diseases as well should Contact this great herbal doctor via his Email : Priestazibasolutioncenter@gmail.com and WhatsApp Him on +2348100368288
ReplyDeleteI just want the whole world to know about this spell caster I met
ReplyDeletetwo weeks ago, wisdomspiritualtemple@gmail.com I cannot say everything he has done for me my wife
left me 3 years ago left with my kids I was going through online
when I meant this wonderful man's testimony online I decided to
give it a try and my wife is back to me now and we ar1e happily
married again cause is too much to put in writing all I can say is
thank you very much am very happy .and does alot of spell
including Love Spell
Death Spell
Money Spell
Power Spell
Success Spell
Sickness Spell
Pregnancy Spell
Marriage Spell
Job Spell
Protection Spell
Lottery Spell
Court Case Spell
Luck Spell etc. In case you need his help contact him on this email
address wisdomspiritualtemple@gmail.com he is a good man
thanks.whatsapp number +234813 648 2342
We specialize in the prevention and treatment in Sydney. We are helping people who have been sexually assaulted. Call a sexual health clinic if you need help or advice.
ReplyDeleteFor more information visit here : Alpha Male Clinic
Happiness is all i see now I never thought that I will be cured from HERPES virus again. I have been suffering from a deadly disease (HERPES) for the past 3 years now, I had spent a lot of money going from one places to another, from churches to churches, hospitals have been my home every day residence. Constant checks up have been my hobby not until this faithful day, I was searching through the internet, I saw a testimony on how DR agaba helped someone in curing his HERPES disease, quickly I copied his email which is Dragabasolutionhome@gmail.com just to give him a test I spoke to him, he asked me to do some certain things which I did, he told me that he is going to provide the herbal cure to me, which he did, then he asked me to go for medical checkup after some days after using the herbal cure, behold I was free from the deadly disease, he only asked me to post the testimony through the whole world, faithfully am doing it now, please brothers and sisters, he is great, I owe him in return. if you are having a similar problem just email him on : Dragabasolutionhome@gmail.com or WhatApp: +2349074536486
ReplyDeleteWe specialize in the prevention and treatment in Sydney. We are helping people who have been sexually assaulted. Call a sexual health clinic if you need help or advice.
ReplyDeleteFor more information visit here coaching certificate
this is real take it serious, who will believe that a herb can cure ten
ReplyDeleteyears HIV in my body, i never believe that this will work i have spend a
lot when getting drugs from the hospital to keep me healthy, what i was
waiting for is death because i was broke, one day i hard about this great
man who is well know of HIV and cancer cure, i decided to email him,
unknowingly to me that this will be the end of the HIV aids in my body, he
prepare the herb for me, and give me instruction on how to take it, at the
end of the two week, he told me to go to the hospital for a check up, and i
went, surprisingly after the test the doctor confirm me negative, i thought
it was a joke, i went to other hospital was also negative, then i took my
friend who was also HIV positive to the Dr voodoo after the treatment she
was also confirm negative . He also have the herb to cure cancer please i
want every one with this virus to be free, that is why am dropping his
email address, voodoospelltemple66@gmail.com i want you to email him he
is a great man. the government is also interested in this Dr voodoo
thank you for saving my life, and I promise I will always testify
for your good work you can also add him on whatsApp
+2348140120719
herpes is a serious and recurring disease which can't be cured through drugs or injections by the American doctors but the best way to deal with herpes is by taking natural herbs medicine for it and is only few American doctors that know about this herbal medicine from Dr Akhanene .. I have read about Dr Akhanene the great herbalist doctor from African who can cure disease with his powerful herbal medicine. for the people suffering from the following diseases, Herpes, Cancer, Also,Herpatitis, Diabetes, Hps,Infections ETC should contact him for his herbal medicine because i am a living testimony and i was cured of herpes. Although, i sent him what he requested and he sent me his medicine which i took for 1 weeks and today when i went for test i was tested herpes negative. you can reach him through his Emai drakhanenespellhome@gmail.com.com or whatsapp or call him +2348168714427
ReplyDelete
ReplyDeleteHERPES SIMPLEX VIRUS TYPE 2 CURE ( https://ehiagwinaherbalhom.wixsite.com/home)
I am from USA i was diagnosed with HSV TPYE 2 while I was pregnant .I cried for days. I was devastated and looking up info on it just made it worse. I don't know how long I have had it for. I don't know if my partner has it or if he was the one who transferred it to me. I had one outbreak while pregnant. That's how I found out I had the virus. I started taking and antiviral and took it everyday twice a day to make sure I wasn't able to get an outbreak. I debated whether I wanted to have my daughter natural or c- section. I was terrified. Stress wasn't helping my situation. I ended up having her natural and she is a beautiful healthy 2 month old baby girl. I try not to think about me having the virus because of my depression i couldn't. so I have to focus more on how to get rid of HSV TPYE 2 because i don't want anything to happen to my daughter and thank God she was born fine. i visited different hospital but they gave me list of drugs like acyclovir, Zovirax, and (valacyclovir)Valtrex without without get rid of my virus. one day i decide to surf the internet for cure so i saw a testimony total cure of HERPES by Dr. EHIAGWINA with NATURAL HERBS so i pick up the email Address: ehiagwinaherbalhome@gmail.com and contact he an hour later he reply explain to me how am going to get the herbs and how am going to use it to cure my virus i give a try believe me it work perfectly i used it within seven days now am tested negative i am free from the virus thanks Doc. for so many passing through this contact Dr. EHIAGWINA to help you get rid of the virus. here is his email address: ehiagwinaherbalhome@gmail.com you can also call or add him on whats app +2348162084504 He can as well cure so many diseases like...........
1. ALS
2. HPV
3. SHINGLES
4. CANCER
5. HIV/AIDS
6. BREAST AND PENIS ENLARGEMENT and others diseases i can't remember now. Thanks Doc am so happy today
DO YOU NEED A PERSONAL/BUSINESS/INVESTMENT LOAN? CONTACT US TODAY VIA WhatsApp +19292227023 Email drbenjaminfinance@gmail.com
ReplyDeleteHELLO
Do you find yourself in a bit of trouble with unpaid bills and don’t know which way to go or where to turn? What about finding a reputable Debt Consolidation firm that can assist you in reducing monthly installment so that you will have affordable repayment options as well as room to breathe when it comes to the end of the month and bills need to get paid? Then consider your financial problems over. We Offer LOANS from $3,000.00 Min to $30,000,000.00 Max with a low interest rate of 2% and loan duration of 1 to 35 years to pay back the loan secure and unsecured. Our loans are well insured for maximum security is our priority, Our leading goal is to help you get the services you deserve, Our program is the quickest way to get what you need in a snap. Kindly reduce your payments to ease the strain on your monthly expenses
NO MATTER YOUR CREDIT SCORE. GET YOUR INSTANT LOAN APPROVAL 100% GUARANTEED TODAY VIA WhatsApp:+19292227023 Email: drbenjaminfinance@gmail.com