Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE) is an acquired inflammatory uveitis that belongs to the heterogenous group of white dot syndromes in which light-coloured (yellowish-white )lesions begin to form in the macular area f retina.Early in the course of the disease, the lesions cause acute and marked vision loss (if it interferes with the optic nerve) that ranges from mild to severe but usually transient in nature. Classified as an inflammatory disorder that is usually bilateral and acute in onset but self-limiting. The lesions leave behind some pigmentation, but visual acuity eventually improves even without any treatment (providing scaring doesn't interfering with optic nerve).
It occurs more commonly in females, and is more likely to affect a persons between 20â€"30 years of ages but has been seen in people 16 to 40 years of age . It is known to occur after or concurrently with a systemic infection (but not always), showing that it is related generally to an altered immune system. Recurrent episodes can happen, but is extremely rare.
Etiology
The aetiology of the inflammation remains unknown, with various theories of it occurring as an autoimmune response to a mild infection, or the possibility of it being viral because of the preceding flu-like illness that generally accompanies it. It is usually associated with HLA-B7 and HLA-DR2
Presentation
The onset of ocular symptoms are usually preceded by episode of viral or flu-like symptoms such as fever, cough or sore throat (however this is not always the case). Patients can typically present erithema nadosum, livido reticularus, bilateral uveitis, and sudden onset of marked visual loss associated with the appearance of multiple lesions in the retina. These lesions may be colored from grey-white to cream-shaded yellow. Other symptoms include scotomata and photopsia. In weeks to a month times the lesions begin to clear and disappear (with prednisone) leaving behind areas of retinal pigment epithelial atrophy and diffuse fine pigmentation (scaring) . Rarely choroidal neovascularization occur as a late onset complication.
Diagnosis
Diagnosis is made by an ophthalmologist during eye examination. Further tests such as Fluorescein angiography and/or lumbar puncture is usually performed to confirm the diagnosis. Similar auto immune disorder that can be confused with APMPPE, is neuro sarcoidosis.
Treatment
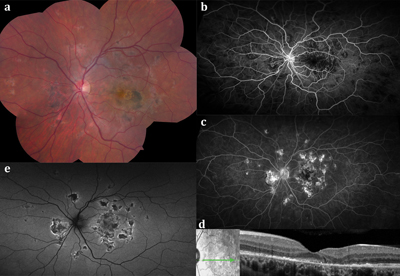
Due to self-limited nature of the disease treatment is generally not required. In the case where lesions appear to be interfering or boarding the optic nerve, methyl prednisone is prescribed.
Prognosis
Vision does improve in almost all cases. Rarely a patient may suffer permanent visual loss associated with lesion on optic nerve.
Rarely, coexisting vasculitis may cause neurological complications. These occurrences can start with mild headaches that steadily increase in pain and threshold and include attacks of dysesthesia. It has been found that this type of deterioration happens usually if the lesions involve the fovea. This is not always the case. I presented with no headache, just fell, and had right hand side weakness, upon presentation to hospital still no head ache but also had slurred speech. This followed another cva (cerebral vascular accident) with reoccurring Tia's. The risk associated with have cva associated with APMPPE is extremely rare.
0 comments:
Post a Comment