Inclusion body myositis (IBM) is an inflammatory muscle disease, characterized by slowly progressive weakness and wasting of both distal and proximal muscles, most apparent in the muscles of the arms and legs. There are two types: sporadic inclusion body myositis (sIBM) and hereditary inclusion body myopathy (hIBM).
In sporadic inclusion body myositis [MY-oh-sigh-tis] muscle, two processes, one autoimmune and the other degenerative, appear to occur in the muscle cells in parallel. The inflammation aspect is characterized by the cloning of T cells that appear to be driven by specific antigens to invade muscle fibers. The degeneration aspect is characterized by the appearance of holes in the muscle cell vacuoles, deposits of abnormal proteins within the cells and in filamentous inclusions (hence the name inclusion body myositis).
sIBM is a rare yet increasingly prevalent disease, being the most common cause of inflammatory myopathy in the over 50s; the most recent research, done in Australia, indicates that the incidence of IBM varies and is different in different populations and different ethnic groups. The authors found that the current prevalence was 14.9 per million in the overall population, with a prevalence of 51.3 per million population in people over 50 years of age. As seen in these numbers, sIBM is an age-related disease â€" its incidence increases with age and symptoms usually begin after 50 years of age. It is the most common acquired muscle disorder seen in people over 50, although about 20% of cases display symptoms before the age of 50. Weakness comes on slowly (over months or years) and progresses steadily and usually leads to severe weakness and wasting of arm and leg muscles. It is slightly more common in men than women. Patients may become unable to perform daily living activities and most require assistive devices within 5 to 10 years of symptom onset. sIBM is not considered a fatal disorder â€" barring complications, all things being equal, sIBM will not kill (but the risk of serious injury due to falls is increased). One common and potentially fatal complication is dysphagia. There is no effective treatment for the disease.
Classification
- The common type is sIBM (sporadic Inclusion Body Myositis): it strikes individuals apparently at random.
- There is a type that has been observed in multiple siblings in the same generation in several families: termed familial inflammatory sIBM, but it is not passed on from generation to generation.
- There are also several very rare forms of hereditary inclusion body myopathy (hIBM) that are linked to specific genetic defects and that are passed on from generation to generation, each inherited in different ways. See hereditary inclusion body myopathy.
Signs and symptoms
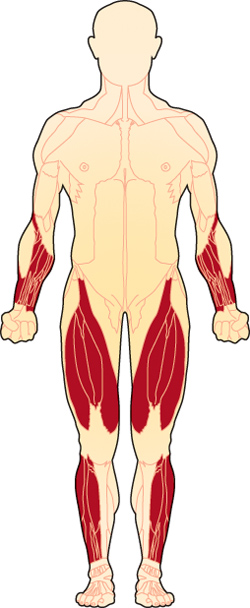
How sIBM affects individuals is quite variable as is the age of onset (which generally varies from the forties upwards). Because sIBM affects different people in different ways and at different rates, there is no "textbook case."
Eventually, sIBM results in general, progressive muscle weakness. The muscles in the thighs called the quadriceps and the muscles in the arms that control finger flexionâ€"making a fistâ€"are usually affected early on. Common early symptoms include frequent tripping and falling, weakness going up stairs and trouble manipulating the fingersâ€"turning doorknobs, gripping keys, etc. Foot drop in one or both feet has been a symptom of IBM and advanced stages of polymyositis (PM).
During the course of the illness, the patient's mobility is progressively restricted as it becomes hard for him or her to bend down, reach for things, walk quickly and so on. Many patients say they have balance problems and fall easily, as the muscles cannot compensate for an off-balanced posture. Because sIBM makes the leg muscles weak and unstable, patients are very vulnerable to serious injury from tripping or falling down. Although pain has not been traditionally part of the "textbook" description, many patients report severe muscle pain, especially in the thighs.
When present, dysphagia is a progressive condition in patients with inclusion body myositis and often leads to death from aspiration pneumonia. Dysphagia is present in from 40 to 85% of IBM cases.
It is also important to note that IBM can result in diminished aerobic capacity. This decline (in aerobic capacity) is most likely a consequence of the sedentary lifestyle that is often associated with the symptoms of IBM (i.e. progressive muscle weakness, decreased mobility, and increased level of fatigue). Therefore, one focus of treatment should be the improvement of aerobic capacity.
Patients with sIBM usually eventually need to resort to a cane or a walker and in most cases, a wheelchair eventually becomes a necessity.
From a recent article: "The progressive course of s-IBM leads slowly to severe disability. Finger functions can become very impaired, such as for manipulating pens, keys, buttons, and zippers, pulling handles, and firmly grasping handshakes. Arising from a chair becomes difficult. Walking becomes more precarious. Sudden falls, sometimes resulting in major injury to the skull or other bones, can occur, even from walking on minimally-irregular ground or from other minor imbalances outside or in the home, due to weakness of quadriceps and gluteus muscles depriving the patient of automatic posture maintenance. A foot-drop can increase the likelihood of tripping. Dysphagia can occur, usually caused by upper esophageal constriction that often can be symptomatically improved, for several months to years, by bougie dilation per a GI or ENT physician. Respiratory muscle weakness can sometimes eventuate."
Causes
The causes of sIBM are currently unknown, though it is likely that it results from the interaction of a number of factors, both genetic and environmental. The understanding of sIBM is slowly maturing and evolving.
Currently, there are two major theories about how sIBM is caused:
1) Some researchers (e.g., Dalakas) advocate the theory that the inflammation-immune reaction, caused by an unknown trigger â€" likely an undiscovered virus or an autoimmune disorder, is the primary, proximal cause of sIBM and that the degeneration of muscle fibres and protein abnormalities are secondary features.
Despite the arguments "in favor of an adaptive immune response in s-IBM, a purely autoimmune hypothesis for s-IBM is untenable because of the disease's resistance to most immunotherapy."
2) Some researchers (e.g., Engel and Askanas) advocate the theory that sIBM is a degenerative disorder related to aging of the muscle fibres and that abnormal, potentially pathogenic protein accumulations in myofibers play a key causative role in s-IBM (apparently before the immune system comes into play). This theory emphasizes the abnormal intracellular accumulation of many proteins, protein aggregation and misfolding, proteosome inhibition, and endoplasmic reticulum (ER) stress.
A recent review by Greenberg (2009) discusses the "limitations in the beta-amyloid-mediated theory of IBM myofiber injury,"
Dalakas (2006) said: "we can say that two processes, one autoimmune and the other degenerative, occur in the muscle cells in parallel."
Dalakas (2006) suggested that a chain of events causes IBMâ€"some sort of virus, likely a retrovirus, triggers the cloning of T cells. These T cells appear to be driven by specific antigens to invade muscle fibers. In people with sIBM, the muscle cells display “flags†telling the immune system that they are infected or damaged (the muscles ubiquitously express MHC class I antigens) and this immune process leads to the death of muscle cells. The chronic stimulation of these antigens also causes stress inside the muscle cell in the endoplasmic reticulum (ER) and this ER stress may be enough to cause a self-sustaining T cell response (even after a virus has dissipated). In addition, this ER stress may cause the misfolding of protein. The ER is in charge of processing and folding molecules carrying antigens. In IBM, muscle fibers are overloaded with these major histocompatibility complex (MHC) molecules that carry the antigen protein pieces, leading to more ER stress and more protein misfolding.
A self-sustaining T cell response would make sIBM a type of autoimmune disorder. One confusing aspect is that medications that lower the immune response do not improve sIBM symptoms, as would be expected in the case of an autoimmune disorder.
When studied carefully, it has not been impossible to detect an ongoing viral infection in the muscles. One theory is that a chronic viral infection might be the initial triggering factor setting IBM in motion. There have been a handful of IBM casesâ€"about 15 or soâ€"that have shown clear evidence of a virus called HTLV-1. This is a complex virus that can cause leukemia, but in most cases it lies dormant and people end up being lifelong carriers of the virus. It is too early to say that this is the particular virus directly involved in causing IBM. The Dalakas article says that the best evidence points towards a connection with some type of retrovirus and that a retroviral infection combined with immune recognition of the retrovirus is enough to trigger the inflammation process.
As mentioned above, in the past, some researchers have suggested that it is the protein changes that are primary and that precede or trigger the abnormal immune response. From an article by Askanas and Engel: "Two hypotheses predominate regarding the key pathogenic mechanisms involved in s-IBM: an amyloid-beta-related degenerative process and an immune dysregulation. Ultimately, both may be considered important, and their possible interrelationship may be clarified. An intriguing feature is the accumulation within s-IBM muscle fibers of amyloid-beta (Ab), phosphorylated tau protein, and at least 20 other proteins that are also accumulated in the brain of Alzheimer's disease patients. In the s-IBM muscle fibers, there is evidence of misfolding of proteins, pathologic proteinaceous inclusions including aggresomes, abnormalities of the two protein-disposal systems involving the ubiquitin proteasome pathway and the lysosomes, mitochondrial dysfunctions, and oxidative stress. The pronounced T-cell inflammation can be striking, and it is characterized by activated, antigen-driven, cytotoxic CD8+ T-cells.
- amyloid protein
- The hypothesis that beta amyloid protein is key to IBM has been supported in a mouse model using an Aβ vaccine that was found to be effective against inclusion body myositis in mouse models. Although this vaccine is likely not safe for human use, it still shows that attacking Aβ has efficacy in mice against IBM.
- Following up on earlier leads, the Greenberg group report finding that the protein TDP-43 is a very prominent and highly sensitive and specific feature of IBM. This protein is normally found within the nucleus but in IBM is found in the cytoplasm of the cell. This important advance should help develop a new screening technique for IBM and may provide clues in terms of a therapeutic approach
Genetic aspects of sIBM
sIBM is not inherited and is not passed on to the children of IBM patients. There are genetic features that do not directly cause IBM but that appear to predispose a person to getting IBM â€" having this particular combination of genes increases one's susceptibility to getting IBM. Some 67% of IBM patients have a particular combination of human leukocyte antigen genes in a section of the 8.1 ancestral haplotype in the center of the MHC class II region. sIBM is not passed on from generation to generation, although the susceptibility region of genes may be.
There are also several very rare forms of hereditary inclusion body myopathy (myopathies) that are linked to specific genetic defects and that are passed on from generation to generation. Because these forms do not show inflammation, they are classified as myopathies and not myositis types. Because they do not display inflammation as a primary symptom, they may in fact be similar, but different diseases to sporadic inclusion body myositis. There are several different types, each inherited in different ways. See hereditary inclusion body myopathy.
A 2007 review that summarized current understanding of the contribution of genetic susceptibility factors to the development of sIBM concluded there is no indication that the genes responsible for the familial or hereditary conditions are involved in sIBM.
Differential diagnosis

IBM is often initially misdiagnosed as polymyositis. A course of prednisone is typically completed with no improvement and eventually sIBM is confirmed. sIBM weakness comes on over months or years and progresses steadily, whereas polymyositis has an onset of weeks or months. Other forms of muscular dystrophy (e.g. limb girdle) must be considered as well.
Diagnosis
Elevated creatine kinase CK levels (at most ~10 times normal) are typical in sIBM but patients can also present with normal CK levels. Electromyography (EMG) studies usually display abnormalities. Muscle biopsy may display several common findings including; inflammatory cells invading muscle cells, vacuolar degeneration, inclusions or plaques of abnormal proteins. sIBM is a challenge to the pathologist and even with a biopsy, diagnosis can be ambiguous.
Treatment
There is no standard course of treatment to slow or stop the progression of the disease. sIBM patients do not reliably respond to the anti-inflammatory, immunosuppressant, or immunomodulatory drugs that have been tried. Management is symptomatic. Prevention of falls is an important consideration. Specialized exercise therapy may supplement treatment to enhance quality of life. Physical therapy is recommended to teach the patient a home exercise program, to teach how to compensate during mobility-gait training with an assistive device, transfers and bed mobility.
Other related disorders
When sIBM was originally described, the major feature noted was muscle inflammation. Two other disorders were also known to display muscle inflammation, and sIBM was classified along with them. They are dermatomyositis (DM) and polymyositis (PM) and all three illnesses were called idiopathic (of unknown origin) myositis or inflammatory myopathies.
It appears that sIBM and polymyositis share some common features, especially the initial sequence of immune system activation, however, polmyositis comes on over weeks or months, does not display the subsequent muscle degeneration and protein abnormalities as seen in IBM, and as well, polymyositis tends to respond well to treatments, IBM does not. IBM is often confused with (misdiagnosed as) polymyositis. Polymyositis that does not respond to treatment is likely IBM.
Dermatomyositis shares a number of similar physical symptoms and histopathological traits as polymyositis, but exhibits a skin rash not seen in polymyositis or sIBM. It may have different root causes unrelated to either polymyositis or sIBM.
References
External links
- Information and links to resources by Bill Tillier
- GeneReview/NIH/UW entry on Inclusion Body Myopathy 2
- Polymyositis and Dermatomyositis Discussion group
- Information from The Myositis Association





An herbal product, Meyseton is formulated by the Herbs Solutions By Nature to treat this serious condition, Inclusion Body Myositis. This product is purely made up of herbs after detailed research and does not produce any unwanted effects.
ReplyDeletehttp://www.herbs-solutions-by-nature.com/Inclusion-Body-Myositis.php
The particular Inclusion Body Myositis Treatment recommended by your expert will rely on the seriousness and kind of issues the nearness of other medical conditions and antagonistic responses to previous treatment.
ReplyDeleteHerbal Treatment for Inclusion Body Myositis also read about the Symptoms, Causes and Diagnosis. Natural Herbal Treatment for Inclusion Body Myositis with Herbal Product Meyseton Natural Supplement for common inflammatory muscle disease. Fight with the Symptoms of Inclusion Body Myositis Naturally.
ReplyDeleteHerbal Treatment for Inclusion Body Myositis also read about the Symptoms, Causes and Diagnosis. Natural Herbal Treatment for Inclusion Body Myositis with Herbal Product Meyseton Natural Supplement for common inflammatory muscle disease. Fight with the Symptoms of Inclusion Body Myositis Naturally.
ReplyDeleteI'm 61 years old, I contracted hpv in 2011' I has be taking lot treatment for it and some months ago the wart stated coming out seriously, I used lot recommendation because there was lot warts around my anus and was so embarrassed. but today I'm totally happy I got the virus eliminated by using natural treatment from Dr Onokun herbal center after his treatment I got cured. all the warts went away' seriously believed Dr Onokun he have the cure for human papillomavirus because he has eliminated hpv been in my body since 2011, Dr Onokun make it possible for me. Here is Dr Onokun email to reach him: Dronokunherbalcure@gmail.com he is welled capable of curing terrible diseases.
ReplyDeleteHappiness is all i see now I never thought that I will live on
ReplyDeleteearth before the year runs out. I have been suffering from a
deadly disease (Herpes) for the past 3 years now; I had spent
a lot of money going from one places to another, from
churches to churches, hospitals have been my home every day
residence. Constant checks up have been my hobby not until
this faithful day, I was searching through the internet, I saw a
testimony on how pp him +2348154637647 Dr Lucky, helped
someone in curing his Herpes disease, quickly I copied his
email which is (drluckyherbalcure@gmail.com) just to give
him a test I spoke to him, he asked me to do some certain
things which I did, he told me that he is going to provide the
herbal cure to me, which he did, then he asked me to go for
medical checkup after some days after using the herbal cure, I
was free from the deadly disease, he only asked me to post
the testimony through the whole world, faithfully am doing it
now, please brothers and sisters, he is great, I owe him in
return. if you are having a similar problem just email him on
(drluckyherbalcure@gmail.com) or Call him or WhatsApp him
+2348154637647
I’m here to testify about what DR. ISIBOR did for me. I have been suffering from (GENITAL HERPES VIRUS) disease for the past 3 years and had constant pain and inching, especially in my private part. During the first year, I had faith in God that i would be cured someday.This disease started circulating all over my body and I have been taking treatment from my doctor, few weeks ago I came across a testimony of Rose Smith on the internet testifying about a Man called DR. ISIBOR on how he cured her from 7 years HSV 2. And she also gave the email address of this man, advise anybody to contact him for help on any kind of diseases that he would be of help, so I emailed him telling him about my (HSV 2) he told me not to worry that I was going to be cured!! Well, I never doubted him I have faith he can cure me too,, DR. ISIBOR prepared and sent me Healing Oil, Soap, roots and herbs which I took. In the first one week, I started experiencing changes all over me, after four weeks of using his Roots/ Herbs, Oil and Soap, I was totally cured. no more inching , pain on me anymore as DR. ISIBOR assured me. After some time I went to my doctor to do another test behold the result came out negative. So friends my advise is if you have such disease or know anyone who suffers from it or any other disease like HPV, HIV, ALS, CANCER etc. you can contact DR. ISIBOR for help via email} drisiborspellhome@gmail.com or call +2348107855231
ReplyDeleteCan't still believe that i got cured from Genital Herpes through herbal treatment from Dr LUCKY who I met through the internet, I actually couldn't believe it at first because it sounded impossible to me knowing how far I have gone just to get rid of it. Dr LUCKY send me his medicine which I took as instructed and here I am living a happy life once again, a big thanks to Dr LUCKY , I am sure there are many herbal doctors out there but Dr LUCKY did it for me, contact him on Email him; { drluckyherbalcure@gmail.com }
ReplyDeleteI’m here to testify about what DR. ISIBOR did for me. I have been suffering from (GENITAL HERPES VIRUS) disease for the past 3 years and had constant pain and inching, especially in my private part. During the first year, I had faith in God that i would be cured someday.This disease started circulating all over my body and I have been taking treatment from my doctor, few weeks ago I came across a testimony of Rose Smith on the internet testifying about a Man called DR. ISIBOR on how he cured her from 7 years HSV 2. And she also gave the email address of this man, advise anybody to contact him for help on any kind of diseases that he would be of help, so I emailed him telling him about my (HSV 2) he told me not to worry that I was going to be cured!! Well, I never doubted him I have faith he can cure me too,, DR. ISIBOR prepared and sent me Healing Oil, Soap, roots and herbs which I took. In the first one week, I started experiencing changes all over me, after four weeks of using his Roots/ Herbs, Oil and Soap, I was totally cured. no more inching , pain on me anymore as DR. ISIBOR assured me. After some time I went to my doctor to do another test behold the result came out negative. So friends my advise is if you have such disease or know anyone who suffers from it or any other disease like HPV, HIV, ALS, CANCER etc. you can contact DR. ISIBOR for help via email} {drisiborspellhome@gmail.com} you can also contact him on WhatsApp +2348107855231
ReplyDeleteAm so grateful Dr Moses anabic herbal home whom made me have an healthy life again,I suffered from herpes virus for 4years and it affected my sexual life I had taken several medications just to get cured but all remained the same. Fortunately I was directed to call dr Moses anabic whom cured me from herpes virus totally with his supernatural herbs .now am living healthy again,he cures all kinds of ailments like cancer, hiv aids,syphilis,chlamydia,fibroid,prematured ejaculation.
ReplyDeleteAny patients can contact him through his personal email address via MOSESANABIC@GMAIL.COM or call/chat with +2348100661264.
I read that Post and got it fine and informative. suplemento barato
ReplyDeleteherpes is a serious and recurring disease which can't be cured through drugs or injections by the American doctors but the best way to deal with herpes is by taking natural herbs medicine for it and is only few American doctors that know about this herbal medicine from Dr Akhanene .. I have read about Dr Akhanene the great herbalist doctor from African who can cure disease with his powerful herbal medicine. for the people suffering from the following diseases, Herpes, Cancer, Also,Herpatitis, Diabetes, Hps,Infections ETC should contact him for his herbal medicine because i am a living testimony and i was cured of herpes. Although, i sent him what he requested and he sent me his medicine which i took for 1 weeks and today when i went for test i was tested herpes negative. you can reach him through his Emai drakhanenespellhome@gmail.com.com or whatsapp or call him +2348168714427
ReplyDeleteI'm very excited to inform everyone that I'm completely cured from my HPV recently. I used Dr Onokun herbal medicine, it's really help during my outbreaks but I totally got cured! from my HPV with a strong and active herbal medicine ordered from a powerful herbalist Dr onokun Contact him via CAll/Whats app +1 208 425 7799 or visit his Facebook page: https://www.facebook.com/naturaltreatmentcenter1
ReplyDelete
ReplyDeleteEliminate herpes forever...........................
This herbal Doctor has a cure to herpes virus,
certainly the best online…
Thank you!! for saving my life
Email_______________R.buckler11(@) gmail . com